Advertisement
Review Series
Open Access | ![]() 10.1172/JCI204551
10.1172/JCI204551
Emerging roles of the cGAS/STING pathway in cardiovascular diseases
Wataru Saitoh,1 Yasutomi Higashikuni,1,2,3,4 Oyunbileg Bavuu,1 Masataka Sata,5 and Daiju Fukuda1
1Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, Osaka, Japan.
2Division of Cardiovascular and Genetic Research, Center for Molecular Medicine, and
3Department of Cardiovascular Medicine, Jichi Medical University, Tochigi, Japan.
4Department of Cardiovascular Medicine, The University of Tokyo, Tokyo, Japan.
5Department of Cardiovascular Medicine, Tokushima University Graduate School, Tokushima, Japan.
Address correspondence to: Daiju Fukuda or Yasutomi Higashikuni, Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka, 545-8585, Japan Phone: 81.6.6645.3801; Email: daiju.fukuda@omu.ac.jp (DF); yasutomi.higashikuni@omu.ac.jp (YH).
Authorship note: WS, YH, and OB contributed equally to this work.
Find articles by Saitoh, W. in: PubMed | Google Scholar
1Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, Osaka, Japan.
2Division of Cardiovascular and Genetic Research, Center for Molecular Medicine, and
3Department of Cardiovascular Medicine, Jichi Medical University, Tochigi, Japan.
4Department of Cardiovascular Medicine, The University of Tokyo, Tokyo, Japan.
5Department of Cardiovascular Medicine, Tokushima University Graduate School, Tokushima, Japan.
Address correspondence to: Daiju Fukuda or Yasutomi Higashikuni, Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka, 545-8585, Japan Phone: 81.6.6645.3801; Email: daiju.fukuda@omu.ac.jp (DF); yasutomi.higashikuni@omu.ac.jp (YH).
Authorship note: WS, YH, and OB contributed equally to this work.
Find articles by Higashikuni, Y. in: PubMed | Google Scholar
1Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, Osaka, Japan.
2Division of Cardiovascular and Genetic Research, Center for Molecular Medicine, and
3Department of Cardiovascular Medicine, Jichi Medical University, Tochigi, Japan.
4Department of Cardiovascular Medicine, The University of Tokyo, Tokyo, Japan.
5Department of Cardiovascular Medicine, Tokushima University Graduate School, Tokushima, Japan.
Address correspondence to: Daiju Fukuda or Yasutomi Higashikuni, Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka, 545-8585, Japan Phone: 81.6.6645.3801; Email: daiju.fukuda@omu.ac.jp (DF); yasutomi.higashikuni@omu.ac.jp (YH).
Authorship note: WS, YH, and OB contributed equally to this work.
Find articles by Bavuu, O. in: PubMed | Google Scholar
1Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, Osaka, Japan.
2Division of Cardiovascular and Genetic Research, Center for Molecular Medicine, and
3Department of Cardiovascular Medicine, Jichi Medical University, Tochigi, Japan.
4Department of Cardiovascular Medicine, The University of Tokyo, Tokyo, Japan.
5Department of Cardiovascular Medicine, Tokushima University Graduate School, Tokushima, Japan.
Address correspondence to: Daiju Fukuda or Yasutomi Higashikuni, Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka, 545-8585, Japan Phone: 81.6.6645.3801; Email: daiju.fukuda@omu.ac.jp (DF); yasutomi.higashikuni@omu.ac.jp (YH).
Authorship note: WS, YH, and OB contributed equally to this work.
Find articles by Sata, M. in: PubMed | Google Scholar
1Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, Osaka, Japan.
2Division of Cardiovascular and Genetic Research, Center for Molecular Medicine, and
3Department of Cardiovascular Medicine, Jichi Medical University, Tochigi, Japan.
4Department of Cardiovascular Medicine, The University of Tokyo, Tokyo, Japan.
5Department of Cardiovascular Medicine, Tokushima University Graduate School, Tokushima, Japan.
Address correspondence to: Daiju Fukuda or Yasutomi Higashikuni, Department of Cardiovascular Medicine, Osaka Metropolitan University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka, 545-8585, Japan Phone: 81.6.6645.3801; Email: daiju.fukuda@omu.ac.jp (DF); yasutomi.higashikuni@omu.ac.jp (YH).
Authorship note: WS, YH, and OB contributed equally to this work.
Find articles by Fukuda, D. in: PubMed | Google Scholar
Published July 1, 2026 - More info
J Clin Invest. 2026;136(13):e204551. https://doi.org/10.1172/JCI204551.
© 2026 Saitoh et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
-
Introduction
Cardiovascular diseases (CVDs) represent a wide array of disorders affecting the heart and blood vessels, including atherosclerosis, hypertension, pulmonary hypertension (PH), aortic aneurysm and dissection (AAD), myocardial infarction (MI), hypertensive heart disease, valvular heart disease, cardiomyopathy, myocarditis, and atrial fibrillation (AF). Despite progress in medical therapies, CVDs remain the leading cause of mortality and morbidity worldwide (1). The prevalence of CVDs, especially in the aging population, is expected to rise in the next few decades (2). To address this global burden, novel therapeutic strategies need to be developed.
The cyclic GMP-AMP synthase/stimulator of IFN genes (cGAS/STING) pathway is a component of the innate immune system that senses cytosolic double-stranded DNA (dsDNA) to trigger proinflammatory gene expression. Originally identified for its role in host defense against viral and bacterial infection through recognition of non-self-DNA, the cGAS/STING pathway has also been implicated in sterile inflammation by detecting self-DNA released from damaged or stressed cells (Figure 1) (3–6). Here, we describe the emerging roles of the cGAS/STING pathway in the pathophysiology of CVDs and discuss therapeutic strategies targeting the pathway.
 Figure 1
Figure 1The cGAS/STING pathway and cardiovascular diseases. (A) Risk factors associated with cardiovascular disease — such as aging, sex differences, obesity, and smoking — contribute to the accumulation of intracellular nuclear DNA and mtDNA and extracellular cell-free DNA (cfDNA), which activate both canonical and noncanonical cGAS/STING signaling. (B) Activation of the cGAS/STING pathway modulates the function of vascular and cardiac cells, including vascular smooth muscle cells (VSMCs), vascular endothelial cells (ECs), immune cells, cardiomyocytes, and fibroblasts. This causes functional and structural changes of the blood vessels and heart, contributing to the pathophysiology of cardiovascular diseases, including atherosclerosis, hypertension, pulmonary hypertension, aortic aneurysm and dissection, myocardial infarction, myocardial ischemia/reperfusion injury, hypertensive heart disease, valvular heart disease, cardiomyopathy, myocarditis, and atrial fibrillation.

-
cGAS/STING in the pathophysiology of vascular diseases
Atherosclerosis. Atherosclerosis is the thickening and hardening of the medium- and large-sized arteries due to a buildup of plaque in the subendothelium that consists of cholesterol, fatty acids, calcium, fibrin, blood cells, cellular waste products, and other substances in the blood. A large body of evidence now indicates that inflammation fundamentally contributes to the pathogenesis of atherosclerosis and accompanying thromboembolic events due to plaque rupture or erosion and that the cGAS/STING pathway plays important roles in producing that inflammation (Figure 1 and Tables 1 and 2). Upregulation of STING is observed in human atherosclerotic lesions (Table 1) (31, 32). In a mouse model of diabetes, genetic deletion and pharmacologic inhibition of STING ameliorate aortic endothelial dysfunction (33). Genetic disruption of IRF3 in Apoe–/– mice resulted in reduced lipid burden and macrophage infiltration in atherosclerotic plaques (34).
In the arterial walls, the cGAS/STING pathway can be activated in various cell types, including vascular endothelial cells (ECs), vascular smooth muscle cells (VSMCs) and immune cells. Dong et al. reported that STING was upregulated in vascular ECs in Apoe–/– mice fed a high-fat diet, and EC-specific deletion of STING in high-fat diet–fed Apoe–/– mice resulted in a meaningful reduction in atherosclerotic plaques (32). Mechanistically, mtDNA, which is released into the cytoplasm of vascular ECs in response to low oscillatory shear stress, was suggested to activate the cGAS/STING pathway, leading to EC senescence and dysfunction and promoting atherogenesis. Oxidized LDL also induces mtDNA release in vascular ECs in Apoe–/– mice fed a Western diet, further contributing to STING activation and atherogenesis (35). Interestingly, activation of the cGAS/STING pathway by mtDNA was reported to contribute to NLRP3 inflammasome activation and subsequent pyroptosis in vascular ECs (36, 37). In vitro, treatment of human umbilical vein ECs with mtDNA or cGAMP increased inflammatory molecule expression and decreased endothelial NOS phosphorylation, partially through the cGAS/STING pathway (33).
Cigarette smoke extract can induce DNA damage in both the nucleus and mitochondria in human ECs, leading to IL-6 upregulation through activation of the cGAS/STING pathway (38). In addition, the cGAS/STING pathway was shown to activate the noncanonical effector PKR-like ER kinase (PERK), which causes vascular EC dysfunction through epigenetic alterations (39). Furthermore, Liu et al. demonstrated that IRF3 upregulation in vascular ECs contributes to plaque vulnerability through adhesion molecule induction in Apoe–/– mice (34). Collectively, these findings indicate the diverse contribution of the cGAS/STING pathway to vascular EC function. However, it is important to note that ECs are heterogenous, with different populations having differing roles in atherosclerosis (40), and the role of the cGAS/STING pathway in EC heterogeneity remains to be elucidated. In addition, the effect of the cGAS/STING pathway on vascular ECs in the peripheral arteries and small vessels needs to be clarified.
A role for the cGAS/STING pathway in atherosclerotic macrophages has also been reported. In Apoe–/– mice fed a Western diet, genetic deletion of STING ameliorated atherosclerosis, with decreased macrophage infiltration and proinflammatory cytokine expression in the aorta, whereas restoration of STING expression in bone marrow–derived cells in Sting–/–;Apoe–/– mice promoted atherogenesis with increased proinflammatory cytokine expression and TBK1 phosphorylation in the aorta (31). The release of mtDNA and subsequent activation of the cGAS/STING pathway have also been reported in oxidized LDL-treated macrophages (41, 42).
In VSMCs, the cGAS/STING pathway is implicated in senescence. Senescent VSMCs produce less collagen and more proinflammatory cytokines, which results in a thin fibrous cap and immune cell infiltration and promotes plaque vulnerability (43, 44). Bi et al. reported that, in Apoe–/– mice with chronic kidney disease, mtDNA released from damaged mitochondria activates the cGAS/STING pathway, resulting in IFN-I production, which causes VSMC senescence and phenotypic switching and promotes plaque vulnerability (43). Uryga et al. demonstrated that telomere-derived damaged DNA induces cellular senescence and proinflammatory cytokine expression in VSMCs through the cGAS/STING pathway, which leads to accelerated atherogenic changes after vascular injury (44).
Hypertension. Hypertension is the most prevalent CVD, in which systemic BP is persistently elevated. Mechanisms of hypertension include high-salt intake, activation of the renin-angiotensin-aldosterone system, increased sympathetic drive, and impaired nitric oxide bioavailability. Accumulating evidence implicates vascular inflammation in the initiation and maintenance of hypertension (45).
A recent study highlighted an important role of the cGAS/STING pathway in the development of hypertension (Figure 1 and Table 3) (46). Li et al. demonstrated that, in mice expressing a human risk variant of apolipoprotein L1 associated with hypertension, STING activation in vascular ECs was associated with hypertension, which promoted endothelin 1 production (46). EC-specific STING deletion markedly attenuated hypertension and subsequent vascular remodeling with suppressed endothelin 1 expression, demonstrating a causal role of STING activation in hypertension.
 Table 3
Table 3Emerging roles of the cGAS/STING pathway in hypertension, hypertensive heart disease, and pulmonary hypertension
Chronobiological rhythms regulate BP and its variability (47, 48), and disruption of those rhythms has been linked to the pathophysiology of hypertension (47, 48). A recent study reported that disruption of chronobiological rhythm activates the cGAS/STING pathway and causes cell senescence in macrophages (49). Since innate immune cells, including macrophages, have been implicated in the pathophysiology of hypertension (50, 51), the cGAS/STING pathway might link disrupted chronobiological rhythms to hypertension. Further studies are needed to clarify the role of the cGAS/STING pathway in circadian control of BP and its variability.
PH. PH is a condition of high BP in the arteries of the lungs (52). Pham et al. observed STING upregulation in the lung of patients with PH (Table 1) (53, 54). In rodent models of PH, genetic disruption or pharmacologic inhibition of STING has been shown to improve pulmonary vascular remodeling and hypertension (Table 3) (54–58). Mechanistically, the cGAS/STING pathway induces VSMC senescence in the pulmonary arteries through NF-κB activation (54–57). Wu et al. showed that STING activates the NLRP3 inflammasome in macrophages to induce proliferation of VSMCs through proinflammatory cytokine production (59). In addition, the cGAS/STING pathway is implicated in immune cell dysfunction in PH. Pham et al. demonstrated that genetic deletion or pharmacologic inhibition of STING increases VEGF in CD11b+ and CD11c+ myeloid cells, which might contribute to protection against PH in an IFN signaling–independent manner (53). Interestingly, Pham et al. also reported that STING expression in myeloid cells might prevent severe PH by suppressing expression of PD-L1, suggesting diverse roles of STING in myeloid cells (54).
AAD. An aortic aneurysm is a dilatation of the aorta to greater than 1.5 times the original size due to structural weakening of the aortic wall. An aortic dissection is a tear between the intima and media of the aortic wall. AADs are lethal vascular conditions, characterized by progressive VSMC loss and extracellular matrix degradation, which can lead to aortic rupture or severe ischemia due to arterial occlusions. Persistent vascular inflammation has been implicated in the pathogenesis of AAD (60).
Recent studies have demonstrated that the cGAS/STING pathway plays important roles in the underlying mechanisms of AAD (Figure 1 and Table 4). Luo et al. reported marked upregulation of STING, TBK1, and IRF3 expression and phosphorylation in human sporadic AAD tissues (Table 1) (61). In a murine model of sporadic AAD induced by a combination of a high-fat diet and high-dose angiotensin II infusion, STING-deficient mice exhibited marked reductions in aortic enlargement, dissection, and rupture. Nuclear and mitochondrial DNA damage in VSMCs, and the subsequent leakage of DNA to the cytosol and extracellular space, activates STING in neighboring VSMCs and macrophages, leading to apoptotic and necroptotic cell death. Using single-cell transcriptome and transposase-accessible chromatin analyses, Chakraborty et al. revealed that activation of STING and IRF3 induces epigenetic modification and chromatin remodeling in VSMCs in the aorta of angiotensin II–infused mice, which resulted in VSMC phenotypic alterations through suppression of contractile genes and induction of proinflammatory genes (62). These epigenetic and phenotypic changes in VSMCs were prevented in Sting–/– mice.
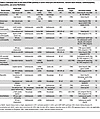 Table 4
Table 4Emerging roles of the cGAS/STING pathway in aortic aneurysm and dissection, valvular heart disease, cardiomyopathy, myocarditis, and atrial fibrillation




-
Targeting cGAS/STING for the treatment of CVDs
Pharmacological inhibitors of the cGAS/STING pathway have been developed, such as cGAS inhibitors, STING inhibitors, and TBK1 inhibitors (Table 5), and have been under clinical investigation in autoimmune disease and cancer (122, 123). VENT-03, an oral cGAS inhibitor, was shown to be safe and well-tolerated in healthy volunteers and is now in a phase IIa trial for the treatment of systemic lupus erythematosus (124). In cancer, nucleotide-based cGAMP analogs, which aim to exploit cGAS/STING-dependent antitumor immunity, have been tested, although trials have not yet shown therapeutic benefit (125). In infectious disease, modulation of cGAS/STING to either augment host defense or mitigate excessive inflammation has been under exploration (126). Collectively, these attempts provide important conceptual and translational frameworks for applying cGAS/STING-targeted therapies to CVDs. Here, we describe evidence of the therapeutic potential of these agents as demonstrated in animal models of CVDs.
 Table 5
Table 5Pharmacological inhibitors of the cGAS/STING pathway for the treatment of cardiovascular diseases
cGAS inhibitors. Three types of cGAS inhibitors have been developed: inhibitors of cGAS enzymatic activity, inhibitors of cGAS binding to dsDNA, and targeted modulators of cGAS by acetylation. RU.521 is a competitive inhibitor of cGAS that binds to the active site of cGAS and inhibits its interaction with ATP and GTP (127). RU.521 has been shown to be effective in the treatment of atherosclerosis, myocardial ischemia/reperfusion injury, and myocarditis in mice (36, 114, 128, 129). X6 and antimalarial drugs, which prevent the formation of the cGAS-dsDNA complex, were shown to have a beneficial effect in murine autoimmune myocarditis (116). Interestingly, the NSAID aspirin acetylates cGAS, which inhibits cGAS activation and improves autoimmune myocarditis in mice (130). Further studies are needed to clarify therapeutic potentials of these agents in human disease.
STING inhibitors. Nitrofuran derivatives, such as C-176, C-178, and H-151, effectively inhibit STING signaling by binding to Cys91, which results in nitroalkylation of STING’s palmitoylation site (131). These compounds have been shown to improve atherosclerosis, MI, myocardial ischemia/reperfusion injury, hypertensive heart disease, cardiomyopathy, and myocarditis in mice (31, 36, 43, 81, 100, 114, 128, 132, 133). Astin C and SN-011 competitively block interaction between STING and cyclic dinucleotides. These compounds showed protective effects against autoimmune myocarditis in mice (134, 135).
TBK1 inhibitors. Several kinase inhibitors, including K252a, dovitinib, and oxindole 91, have been shown to inhibit TBK1 by activity-based screens of compound libraries, although they are not TBK1-specific inhibitors (136). These compounds prevent autophosphorylation of TBK1 and IκB kinase ε (IKKε) at the Ser172 site through hydrogen bond network formation. Amlexanox is another TBK1 inhibitor that prevents its phosphorylation at the Ser366 site, and it also has an inhibitory effect on IKKε. Amlexanox is currently used for the treatment of recurrent aphthous ulcers and bronchial asthma. Several studies have shown that amlexanox mitigates atherosclerosis and AAD in mice (31, 61). Further investigation will be needed to determine the potential of TBK1 inhibitors in the treatment of CVDs.

Article tools
- Download citation information
- Send a comment
- Terms of use
- Standard abbreviations
- Need help? Email the journal
Review Series
The cGAS-STING pathway: DNA sensing in health and disease
-
Interstitial lung disease and the STING pathway
-
Therapeutic targeting of the cGAS-STING pathway in human disease
-
Molecular mechanisms regulating cGAS/STING activation in health and disease
-
Autoinflammatory syndromes of STING and TREX1 dysfunction
-
Expanding roles of cGAS-STING signaling in neuroinflammation
-
Emerging roles of the cGAS/STING pathway in cardiovascular diseases
Metrics
Go to
- Top
- Abstract
- Introduction
- Overview of the cGAS/STING pathway
- DNA recognized by the cGAS/STING pathway
- cGAS/STING in the pathophysiology of vascular diseases
- cGAS/STING in the pathophysiology of heart diseases
- Targeting cGAS/STING for the treatment of CVDs
- Conclusions and remaining questions
- Conflict of interest
- Funding support
- Footnotes
- References
- Version history



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)