Nephrology
Abstract
The inflammatory response resulting from the abnormal accumulation of metabolites has been implicated in the pathogenesis of organ fibrosis; however, its role and underlying mechanisms in renal fibrosis remain unclear. In this study, we observed a negative correlation between fumarate hydratase (FH) expression and the degree of renal fibrosis. Loss of FH function was associated with heightened inflammation and exacerbated tubulointerstitial damage in the kidney. Moreover, FH deficiency aggravated fibrosis in both the liver and lungs. Mechanistically, the depletion of FH in renal tubular cells led to fumarate accumulation. Fumarate directly succinated A-kinase anchoring protein 12 at cysteine 670, thereby diminishing its capacity to inhibit the activity of protein kinase Cζ (PKCζ). This process exacerbated renal inflammation and fibrosis by activating the downstream PKCζ/NF-κB and PKCζ/β-catenin pathways. Additionally, the upregulation of FH through adeno-associated virus 2/9-mediated FH overexpression markedly mitigated renal inflammation and fibrosis. These findings highlighted the important role of fumarate accumulation in the advancement of renal fibrosis, supporting FH as a potential therapeutic target in renal fibrosis.
Authors
Shuai Sun, Xu-yang Yan, Yu-hang Dong, Jian-min You, Zhen-yu Guo, Dong-xue Lv, Shuai-shuai Xie, Rui Hou, Xiang-yu Li, Ju-tao Yu, Xiao-yu Shen, Jie Wei, Zhen-yu Song, Zi-qi Chen, Yun-long Zhu, Xing-xin Xu, Juan Jin, Jia-gen Wen, Hao Wang, Xiao-ming Meng, Wei Wang
Abstract
Autosomal dominant polycystic kidney disease (ADPKD), the leading genetic cause of kidney failure, results from loss-of-function mutations in PKD1, encoding polycystin-1 (PC1). PC1 localizes to the primary cilium. In the absence of PC1, adverse signaling from the primary cilium orchestrates cyst formation, but the biomechanical underpinnings of this cilia-dependent cyst activation (CDCA) remain unclear. Combining tubule-specific orthologous mouse models with a tubule-on-chip platform, we show that PC1 and cilia govern the composition, mechanical properties and shape of the tubular basement membrane (TBM), the principal rigid determinant of tubule geometry. PC1 loss triggers TBM thinning, heparan sulfate enrichment and deformation, leading to distension, preferentially of the distal nephron. These changes are driven by a cilia-dependent transcriptional program, with GLIS2 — a key CDCA effector — participating as a downstream mediator. Reduction of TBM stiffness amplifies Pkd1−/− tubule-on-chip dilation and increases cyst formation in vivo. Conversely, increasing luminal pressure through ureteral obstruction induces disproportionate distension of Pkd1-deficient tubules and triggers an irreversible cystogenic program. Together, these findings establish a TBM-centered biomechanical model of ADPKD in which tubule deformation is governed by both basolateral and luminal mechanical factors, and identify the cilium–TBM axis, operating in part through GLIS2, as a central driver of cystogenesis.
Authors
Manal Mazloum, Brice Lapin, Rushdi Alghamdi, Jessica Vandensteen, Martine Burtin, Pascal Houillier, Lydie Cheval, Gilles Crambert, Vicky Scata, Camille Cohen, Christoph Schell, Michael Rehman, Amandine Aka, Karim Ourahmoun, Rui Benedito, E. Wolfgang Kuehn, Stéphanie Descroix, Tilman Busch, Michael Köttgen, Serge Garbay, Marie-Christine Verpont, Ellie Tang, Brigitte Lelongt, Nicolas Cagnard, Stefan Somlo, Sylvie Coscoy, Fabiola Terzi, Amandine Viau, Frank Bienaimé
Abstract
Renal water reabsorption is classically regulated by vasopressin V2 receptor (V2R) signaling through cyclic AMP and protein kinase A, driving apical accumulation of aquaporin-2 (AQP2). However, collecting duct water handling is also modulated by vasopressin-independent mechanisms. Here, we examined intracellular soluble urate as a vasopressin-independent regulator of AQP2 trafficking. Intracellular urate accumulation in collecting duct cells was mediated by enhanced apical urate uptake via GLUT9b and reduced apical urate efflux through ABCG2, triggering phosphodiesterase-4 activation, reduced cAMP, and downstream AMP-activated protein kinase (AMPK) activation. The resulting AQP2 accumulation at the apical membrane was independent of V2R signaling, required ongoing endocytosis and was associated with features of post-endocytic apical trafficking of internalized AQP2. In vivo ABCG2 inhibition with probenecid increased apical AQP2 abundance and markedly attenuated tolvaptan-induced polyuria in both wild-type and Pkd1RC/RC autosomal dominant polycystic kidney disease (ADPKD) mice in a uricase-independent manner, while preserving tolvaptan’s ADPKD-modifying efficacy. In a Phase 2 trial with tolvaptan-treated ADPKD patients, probenecid reduced urine volume and nocturia frequency. Together, these findings support a vasopressin-independent urate–AMPK–AQP2 pathway that regulates renal water handling and, in a preclinical ADPKD model, can uncouple cyst growth attenuation from the dose-limiting aquaretic effects of V2R antagonism.
Authors
Mohamad Hadla, Jean Marc Mardirossian, Daniel G. Bichet, Abdul Hamid Borghol, Georges Abboud, Ahmad Ghanem, Eduardo N. Chini, Peter C. Harris, Vicente E. Torres, Seth L. Alper, Volker Vallon, Fouad T. Chebib
Abstract
Exonic variants in Apolipoprotein-L1 (G1 and G2) are linked to increased risk of kidney disease as well as kidney transplant rejection. Outside of the association of these prevalent variants with African ancestry, underpinning causal mechanisms for rejection are unknown. We investigated T-cell function using transgenic mice with physiologic expression of wild type (G0-), G1-APOL1 (G1), or G2-APOL1 (G2). Mice with either variant showed greater CD8+T-cell activation with expansion of a central memory (TCM) subset. Stimulated G1-CD8+T-cells showed enhanced proliferation and cytokine production, which reversed with APOL1 inhibition. In MHC-mismatched cardiac transplants, G1-mice demonstrated greater CD8+T-cell infiltration and reduced survival. Bulk transcriptome of G1-CD8+T-cells, and single-cell transcriptome of graft infiltrating TCMs, showed enrichment of canonical T-cell receptor (TCR) pathways including Ca2+-signaling. G1-CD8+T-cells demonstrated baseline ER-Ca2+ depletion followed by sustained increases in cytosolic-Ca2+ upon TCR stimulation. G1-CD8+T-cells were more sensitive to Ca2+ chelation, or store-operated Ca2+ entry inhibition, and were relatively resistant to calcineurin antagonism compared to G0-CD8+T-cells. Analogously, in a kidney transplant cohort, APOL1-variant recipients that had elevated peripheral TCMs before transplantation, developed rejection despite significantly higher tacrolimus levels vs G0/G0 recipients. In summary, we unravel an excitatory mechanism for APOL1 variants in T-cells that causally links them to kidney rejection.
Authors
John Pell, EM Tanvir, Zeguo Sun, Irene Chernova, Anand Reghuvaran, Soichiro Nagata, Mateus T. Guerra, John Choi, Soltan Al Chaar, Hiroki Mizuno, Ke Dong, Xin Tian, Reika Ishibe, Barbara Franchin, Paolo Cravedi, Ashwani Kumar, Gabriel Barsotti, Hongmei Shi, Bony De Kumar, Shinobu Smithson, Wenzhi Song, John Cijiang He, Anita S. Chong, Jordan S. Pober, Stefan Somlo, Ian W. Gibson, Waldemar Popik, Zhongyang Zhang, Joseph Craft, Jamil Azzi, Naoka Murakami, Shuta Ishibe, Peter S. Heeger, Madhav C Menon
Abstract
BACKGROUND. Anti-nephrin autoantibodies have emerged as a putative pathogenic driver in a subset of patients with podocytopathies, including those with post-transplant disease recurrence. METHODS. We measured anti-nephrin autoantibodies in a cohort of 65 patients with podocytopathy associated with steroid-sensitive nephrotic syndrome (n = 39) and steroid-resistant nephrotic syndrome (n = 26), and in 34 patients with post-transplant podocytopathy recurrence. Fourteen patients with membranous nephropathy and 20 healthy volunteers served as controls. ELISA and immunoprecipitation assays were performed to detect anti-nephrin IgG using two different recombinant human nephrin proteins. Immunofluorescence analysis was performed to assess the deposition of IgG and their colocalization with nephrin in renal biopsies. RESULTS. When using murine antigen-based ELISA, the highest positivity was found in healthy volunteers (55%), correlating with levels of circulating natural anti-α-galactose-α-1,3-galactose antibodies. This cross-reactivity was abrogated with recombinant human nephrin expressed in human cells. In this setting, very low prevalence (<5%) of anti-nephrin antibody-positive patients was found in steroid-sensitive and steroid-resistant nephrotic syndrome cohorts and in patients with post-transplant disease recurrence. These frequencies were comparable to healthy volunteers. Using confocal and super-resolution microscopy, only trace amounts of IgM, but no IgG, were found in the glomeruli of analyzed biopsies, which did not colocalize with nephrin. CONCLUSIONS. With the methodology presented here, anti-nephrin reactivity was extremely rare and occurred at comparably low frequencies in healthy controls, native-kidney podocytopathies, and post-transplant disease recurrence. This suggests that these autoantibodies are not inherently disease-specific and may not serve as a broad biomarker across podocytopathies. TRIAL REGISTRATION. ClinicalTrials.gov NCT06334692. FUNDING. Private donation.
Authors
Francesco Pecoraro, Luca Perico, Federica Casiraghi, Paola Rizzo, Matias Trillini, Andrea Angeletti, Manuel Alfredo Podestà, Xhuliana Kajana, Agnese Spennacchio, Marta Todeschini, Marilena Mister, Giuseppe Castellano, Ariela Benigni, Giuseppe Remuzzi
Abstract
Sphingosine-1-phosphate lyase (SPL) insufficiency syndrome (SPLIS) or nephrotic syndrome type 14 (NPHS14), is an autosomal recessive multisystem disorder caused by loss-of-function mutations in SGPL1, encoding the enzyme responsible for the terminal degradation of sphingosine-1-phosphate (S1P). We investigated a patient carrying a previously undescribed c.1084T>A (p.Ser362Thr) SGPL1 variant and analyzed the metabolic and cellular consequences of SPL deficiency using patient fibroblasts, SGPL1-knockout HEK293T cells, and Sgpl1–/– and Sgpl1rosa+fl/fl mice. Metabolic stable isotope labelling revealed that SPL deficiency does not invariably result in S1P accumulation. Instead, SPL-deficient cells maintain near-normal S1P levels through (i) feedback regulation of de novo sphingolipid synthesis via the ORMDL–ceramide axis and (ii) increased diversion of excess ceramides into glycosphingolipids. However, perturbation of sphingolipid homeostasis — either by exogenous sphingolipid load or disruption of compensatory regulation — induces pathological intracellular S1P accumulation. In vivo, Sgpl1–/– mice exhibited pronounced urinary S1P excretion and renal S1P enrichment, accompanied by cytoskeletal disorganization and impaired epithelial morphogenesis. Mechanistically, we identify aberrant Rho–ROCK signaling as a key mediator of S1P-driven cytoskeletal dysregulation. Pharmacological ROCK inhibition with Fasudil mitigated renal cytoskeletal defects in Sgpl1–/– and Sgpl1rosa+fl/fl mice and partially restored epithelial architecture. These findings redefine the metabolic consequences of SPL deficiency and identify S1P-driven Rho–ROCK hyperactivation as a tractable therapeutic target in SPLIS.
Authors
Adam Majcher, Ranjha Khan, Kathrin Buder, Florence Bourquin, Julie D. Saba, Thorsten Hornemann

Abstract
Repetitive injuries are an important trigger of progressive fibrosis. To study if repetitive injuries induce an accelerated profibrotic process, also called “fibrosis-memory,” we established an experimental system with two consecutive, clearly separated insults in a model of renal fibrosis with reversible and irreversible unilateral ureteral obstruction. We found that a preceding fibrotic event of one kidney markedly enhanced subsequent development of fibrosis in the contralateral kidney. Aggravation of fibrosis during the second insult was dependent on memory CD4+ T cells. T cell depletion abrogated the fibrosis-memory effect, while adoptive transfer of memory T cells from fibrotic mice enhanced fibrosis in the recipients. Moreover, IL-3 production by memory CD4+ T cells was essential for aggravation of fibrosis in memory situations. In patients with systemic sclerosis, IL-3 expression by T cells was markedly increased, especially after a long disease duration accompanied by involvement of internal organs. In summary, our data identify IL-3–mediated fibrosis-memory as an important driver of progressive fibrosis.
Authors
Simone Buchtler, Antje Frühauf, Sophia Neumayer, Kathrin Schmidbauer, Yvonne Talke, Frederike Winter-Köhler, Saidou Balam, Karin Landgraf, Claudia Gebhard, Michael Rehli, Florian Volker Schlieckau, Maria Beck, Florian Günther, Martin Fleck, Kerstin Renner, Matthias Mack
Abstract
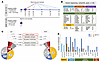
BACKGROUND Kidney stone disease (KSD) affects approximately 10% of the population. While genetic factors are known to play a role in KSD, determining the clinical relevance of rare variants in KSD genes identified in adults remains challenging.METHODS The Swiss Kidney Stone Cohort is a multicenter longitudinal, observational study consisting of kidney stone formers (KSFs) (n = 701) and non-kidney stone formers (NKSFs) (n = 200). Blood and urine samples were collected at enrollment and over 3 years for deep biochemical phenotyping. Results were correlated with rare genetic variants in established KSD genes identified through whole-exome sequencing and classified according to American College of Medical Genetics and Genomics and the Association of Molecular Pathology (ACMG/AMP) criteria.RESULTS Collectively, we found rare (likely) pathogenic (LP/P) variants representing strong KSD risk factors in 6.8% of KSFs, predominantly in genes involved in renal phosphate handling and cystinuria. Detailed biochemical analyses confirmed that KSFs carrying heterozygous LP/P SLC34A3 variants exhibited significant hyperphosphaturia. In contrast, monoallelic LP/P variants in SLC34A1, SLC9A3R1, or CYP24A1, which were also frequent in NKSFs, did not result in the expected biochemical alterations, calling into question their causative role as strong KSD risk factors. In cystinuria, monoallelic SLC7A9 variants represented intermediate risk factors, since they caused biochemical alterations but required additional factors for KSD occurrence, based on frequent LP/P variants in NKSFs. The presence of strong risk factors was associated with higher kidney stone (KS) recurrence over the 3-year observation period, supporting a predictive value for genetic testing.CONCLUSIONS Correlation of genetic findings with thorough biochemical phenotyping and comparison with NKSFs redefines the clinical relevance of variants in KSD genes and has prognostic value.
Authors
Johannes Münch, Jana Petrovska, Joana Figueiro-Silva, Isabel Rubio-Aliaga, Elena M. Cabello, Ivan Ivanovski, Michael Papik, Beatrice Oneda, Daniel G. Fuster, Harald Seeger, Thomas Ernandez, Florian Buchkremer, Gregoire Wuerzner, Nasser A. Dhayat, Alexander Ritter, Stephan Segerer, Beat Roth, Anita Rauch, Pietro Manuel Ferraro, Olivier Bonny, Carsten A. Wagner, Ruxandra Bachmann-Gagescu
Abstract
Background Youth with type 2 diabetes (T2D) and severe obesity face high risk of diabetic kidney disease, which metabolic bariatric surgery (MBS) can mitigate. This study explores structural and molecular changes in kidneys after vertical sleeve gastrectomy (VSG), a form of MBS. Methods Paired analyses, including metabolic profiling, kidney volume assessment, histological evaluation, and single-cell RNA sequencing (scRNAseq) on kidney biopsies from five youth with T2D and obesity pre- and 12 months post-VSG in the IMPROVE-T2D (Impact of Metabolic surgery on Pancreatic, Renal and cardiOVascular hEalth in youth with T2D) cohort. Circulating proteomics with kidney transcriptomics, were linked using data from an independent cohort of youth with obesity, with or without T2D, undergoing MBS in Teen-Longitudinal Assessment of Bariatric Surgery (Teen-LABS, n=64). Results Post-VSG, participants lost weight and had improvements in insulin sensitivity and metabolic parameters. Kidney changes included reduced renal hyperfiltration, total kidney volume, mesangial matrix area, and microalbuminuria. scRNAseq in proximal tubule (PT) and thick ascending limb cells indicated repression of glycolysis, gluconeogenesis, and tricarboxylic acid cycle genes, with upregulation of AMP-activated protein kinase (AMPK) and Forkhead box O3 (FOXO3). Decreased metabolic signaling aligned with reduced ribosomal phosphorylated S6K (pS6K), suggesting attenuated mTORC1 activity. JAK-STAT pathway activation in PT was diminished, correlating with lower circulating ligands from Teen-LABS proteomic data. Conclusion MBS/VSG prompts kidney molecular adaptations, providing potential targets for non-surgical interventions against obesity- and diabetes-associated kidney disease.
Authors
Abhijit S. Naik, Fadhl M. Alakwaa, Viji Nair, Phillip J. McCown, Jennifer A. Schaub, Edgar A. Otto, Rajasree Menon, Francesca Annese, Ye Ji Choi, Hailey E. Hampson, Thomas H. Inge, John Hartman, Sean Eddy, Cathy Smith, Jeffrey B. Hodgin, Ken Inoki, Swayam Prakash Srivastava, Kareem Al-Fagih, Shota Yoshida, Jesse A. Goodrich, Melanie G. Cree, Phoom Narongkiatikhun, Long Yuan, Kalie L. Tommerdahl, Pottumarthi Prasad, Daniël H. van Raalte, Megan M. Kelsey, Justin R. Ryder, Tyler J. Dobbs, Patricia Ladd, Subramaniam Pennathur, Robert G. Nelson, Yusuke Okabayashi, Victor G. Puelles, Jenna Ferrence-Salo, Jeffrey A. Beamish, Frank C. Brosius, Kristen J. Nadeau, Laura Pyle, Matthias Kretzler, Petter Bjornstad

Abstract
The urokinase plasminogen activator receptor (uPAR) is a membrane-bound protein found on the surface of immune cells. Through the action of proteases, uPAR is cleaved to produce several circulating proteins in the bloodstream, including the soluble form suPAR and the fragments D1 and D2D3. Initially studied in the context of infectious diseases and cancer, recent research has revealed roles for suPAR and its related proteins as mediators linking innate immunity to the pathogenesis of kidney and cardiovascular diseases, as well as insulin-dependent diabetes. While these proteins have long been recognized as prognostic biomarkers, growing clinical, experimental, and genetic evidence highlights their active involvement in the onset and progression of these diverse conditions. This Review examines suPAR’s evolution from its discovery as a modulator of innate immunity to its current status as a key driver in chronic kidney and cardiovascular diseases. Furthermore, we explore the molecular mechanisms through which suPAR and D2D3 contribute to multiorgan damage, emphasizing emerging opportunities for therapeutic interventions across interconnected organ systems.
Authors
Jochen Reiser, Salim S. Hayek, Sanja Sever












Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)