Endocrinology
Abstract
Chronic primary pain conditions (CPPCs), such as fibromyalgia and vestibulodynia, affect over 100 million Americans, predominantly women, and pose a substantial healthcare challenge. CPPCs arise from genetic and environmental factors that enhance catecholamine tone, potentially through miRNA dysregulation following catecholamine activation of beta-adrenergic receptors. Here, we identified miR-133a-3p as a biomarker of CPPC status and investigated its functions using in vivo and in vitro approaches. Plasma levels of miR-133a-3p were consistently downregulated in humans with ≥1 CPPC and in rat and mouse models of primary pain. Our data suggest that miR-133a-3p is packaged in extracellular vesicles that are secreted by adipocytes and trafficked to the spinal cord. Activation of adrenergic receptors on white adipocytes resulted in downregulation of miR-133a-3p which negatively regulated pain-related genes in the spinal cord, such as MAP3K3, which is critical for sensory neuron activation. Adipose-specific overexpression of miR-133a-3p in a mouse model of primary pain reversed mechanical hypersensitivity in both sexes. These findings implicate miR-133a-3p dysregulation in primary pain across conditions and species and establish its role in multi-site mechanical hypersensitivity. Further, miR-133a-3p overexpression shows therapeutic potential for the millions of individuals with CPPCs.
Authors
Nathaniel P. Hernandez, Jiegen Chen, Yiling Qian, Xin Zhang, Yaomin Wang, Brittney P. Ciszek, Xianglong Gao, Marguerita E. Klein, Yun-Ling Pai, Mohamad Karaky, Carolina B. Meloto, Francesca Montagna, Matt Kanke, Clair Crewe, Luda Diatchenko, Praveen Sethupathy, Andrea G. Nackley
Abstract
BACKGROUND. In female murine models, one source of inflammation is a menopause-related increase in gut permeability. We examined whether the menopause transition (MT) in women is associated with an increase in markers of gut epithelial dysfunction and gut microbial product translocation, signals of compromised gut epithelial barrier integrity. METHODS. In 964 women, we measured markers of gut epithelial dysfunction (fatty acid binding protein 2, FABP2) and gut microbial antigen translocation (soluble CD14, sCD14) using sera collected before, during and after the MT. Multivariable mixed effects regressions fit piece-wise linear models to repeated FABP2 or sCD14 measures relative to time from final menstrual period (FMP). Covariates were age at FMP, race/ethnicity, and BMI. RESULTS. FABP2 and sCD14 did not change significantly until 2.5 years pre-FMP. At that point, FABP2 began rising; sCD14 began increasing 6 months later. FABP2 and sCD14 peaked 6 and 6.5 years post-FMP, respectively; subsequent levels remained stable. During the ~9-year interval of MT-related gain in gut barrier compromise markers, annual FABP2 and sCD14 increases were 2.6% (95% CI: 1.7 to 3.4%) and 0.8% (95% CI: 0.6 to 1.1%), respectively, among white women with sample-average BMI and age at FMP. FABP2 and sCD14 change rates did not differ significantly by race/ethnicity, BMI, or age at FMP. CONCLUSIONS. The MT is associated with a rise in markers of compromised gut barrier integrity, suggesting that this pathway of inflammation, previously described in animal models, occurs in humans. FUNDING. NIH U01NR004061, U01AG012505, U01AG012535, U01AG012531, U01AG012539, U01AG012546, U01AG012553, U01AG012554, U01AG012495, 5R01AR081794.
Authors
Albert Shieh, Marta Epeldegui, Arun S. Karlamangla, Rheinallt Jones, Roberto Pacifici, Gail A. Greendale

Abstract
Authors
Abdualrahman Mohammed Abdualkader, Xiaobei Li, Yiming Yin, Chenhao Bai, Parisa Pourfarziani, Jiaheng Guan, Sora Kwon, Kyoung-Han Kim, Rami Al Batran

Abstract
Androgen deprivation therapy (ADT), a cornerstone of advanced prostate cancer treatment, effectively suppresses androgen signaling but frequently induces systemic metabolic dysregulation. Here, we delineate an unrecognized intestinal steroid/bile acid regulatory axis that mechanistically links androgen suppression to extratumoral metabolic aberrations. HSD3B1 is the most common inherited link to prostate cancer mortality and mediates its effects by regulating steroid metabolism. Integrated metabolomic profiling of patients undergoing ADT revealed a rapid genotype-associated reduction in circulating bile acids, most pronounced in carriers of the adrenal-permissive HSD3B1 (1245C) allele. Surprisingly, analyses in human intestinal tissue and mechanistic investigations in in vitro models identified the terminal ileum as a unique site of HSD3B1 and SLC10A2 (ASBT) coexpression, where catalytically active 3βHSD1 is transcriptionally governed by liver receptor homolog-1 (LRH-1). Pharmacologic or genetic LRH-1 inhibition coordinately suppressed HSD3B1 and SLC10A2 expression and function, while inducing adaptive HSD11B2 upregulation and enhanced glucocorticoid inactivation. This LRH-1–dependent regulatory program persisted independently of androgen and glucocorticoid receptor signaling under in vitro conditions modeling androgen deprivation. These findings establish LRH-1 as a central integrator of intestinal steroidogenesis and bile acid transport and implicate the LRH-1/HSD3B1/SLC10A2 network as a mechanistic driver of ADT-associated metabolic disturbances and a potential target for therapeutic intervention.
Authors
Nikou Fotouhi, Robert Diaz, Mohammad Alyamani, Yoon-Mi Chung, Gail West, Pranab K. Mukherjee, Alireza Abdshah, Robert A. Burgess, Samreen Jatana, Rana R. McKay, Florian Rieder, Mary-Ellen Taplin, Nima Sharifi

Abstract
Interrupting glucagon signaling decreases gluconeogenesis and the fractional extraction of amino acids by liver from blood, resulting in lower glycemia. The resulting hyperaminoacidemia stimulates α cell proliferation and glucagon secretion via a liver/α cell axis. We hypothesized that α cells detect and respond to circulating amino acids’ levels via a unique amino acid transporter repertoire. We found that Slc7a2/SLC7A2 is the most highly expressed cationic amino acid transporter in α cells, with its expression being 3-fold greater in α than β cells in both mouse and human. Employing cell culture, zebrafish, and knockout mouse models, we found that the cationic amino acid arginine and SLC7A2 are required for α cell proliferation in response to interrupted glucagon signaling. Ex vivo and in vivo assessment of islet function in Slc7a2–/– mice showed decreased arginine-stimulated glucagon and insulin secretion. We found that arginine activation of mTOR signaling and induction of the glutamine transporter SLC38A5 was dependent on SLC7A2, showing that the role of both in α cell proliferation is dependent on arginine transport and SLC7A2. Finally, we identified single nucleotide polymorphisms in SLC7A2 associated with HbA1c. Together, these data indicate a central role for SLC7A2 in amino acid–stimulated α cell proliferation and islet hormone secretion.
Authors
Erick Spears, Jade E. Stanley, Matthew Shou, Linlin Yin, Xuan Li, Chunhua Dai, Amber Bradley, Katelyn Sellick, Greg Poffenberger, Katie C. Coate, Shristi Shrestha, Anna Marie R. Schornack, Taverlyn Shepard, Madushika Wimalarathne, Regina Jenkins, Kyle W. Sloop, Keith T. Wilson, Alan D. Attie, Mark P. Keller, Wenbiao Chen, Alvin C. Powers, E. Danielle Dean
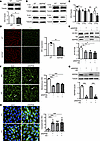
Abstract
Diabetic retinopathy involves early retinal vascular barrier breakdown and pericyte loss, yet the initiating molecular events remain poorly defined. Vascular endothelial cadherin (VE-cadherin), a key regulator of endothelial integrity, is notably reduced in diabetic and prediabetic nucleoside diphosphate kinase B–deficient (NDPKB-deficient) mouse retinas, particularly in the retinal deep capillary layer, and this decline precedes pericyte loss. In vitro, high glucose (HG) and NDPKB deficiency induced VE-cadherin Y685 phosphorylation, promoting its junctional internalization, activating the hexosamine biosynthesis pathway, and increasing angiopoietin 2 (Ang2), resulting in impaired endothelial barrier function and disrupting pericyte attachment. Preventing Y685 phosphorylation through VE-cadherin Y685F mutation blocked these HG- and NDPKB-driven pathological effects. Pharmacological intervention experiments identified protein O-linked β-N-acetyl glucosamine (O-GlcNAc) modification as a mediator of Y685-dependent Ang2 upregulation. In vivo, VE-cadherin Y685F-knockin mice were protected from diabetes- and prediabetes-induced vascular hyperpermeability, exhibited reduced protein O-GlcNAcylation and Ang2 induction, and maintained neuronal function. O-GlcNAc–enriched retinal proteomics further showed that the Y685F mutation restored balanced neurovascular and mitochondrial pathways. These findings highlight the potential of targeting VE-cadherin Y685 phosphorylation as a promising therapeutic approach to maintain retinal vascular integrity and attenuate the pathological progression of diabetic and prediabetic retinopathy.
Authors
Yixin Wang, Hongpeng Huang, Feng Shao, Rachana Eshwaran, Miao Qin, Noor Karim, Yonggang Ren, Gergana Dobreva, Hans-Peter Hammes, Thomas Wieland, Yuxi Feng

Abstract
Authors
Fumihiko Urano, Bess A. Marshall, Stacy Hurst, Amy Robichaux-Viehoever, Saumel Ahmadi, Tamara Hershey, Gregory Van Stavern, Paulina Cruz Bravo, Jennifer Powers Carson, John Pesko, Kelly Fox, Nathalie Erpelding, Camille L. Bedrosian

Abstract
Thyroid hormones (THs [T3 and T4] ) are key regulators of metabolic rate and nutrient metabolism. They are controlled centrally and peripherally in a coordinated manner to elegantly match T3-mediated energy expenditure (EE) with energy availability. Hypothyroidism reduces EE and has long been blamed for obesity; however, emerging evidence suggests that, instead, obesity may drive thyroid dysfunction. Thus, we used a mouse model of diet-induced obesity to determine its direct effects on thyroid histopathology and function, deiodinase activity, and T3 action. Strikingly, overnutrition induced hypothyroidism within 3 weeks. Levels of thyroidal THs and their precursor protein thyroglobulin decreased, and ER stress was induced, indicating that thyroid function was directly impaired. We also observed pronounced histological and vascular expansion in the thyroid. Overnutrition additionally suppressed T4 activation, rendering the mice resistant to T4 and reducing EE. Our findings collectively show that overnutrition deals a double strike to TH biosynthesis and action, despite large efforts to adapt — but, fortunately, thyroid dysfunction in mice can be reversed by weight loss. In humans, BMI correlated with thyroidal vascularization, importantly demonstrating preliminary translatability. These studies lay the groundwork for obesity therapies that tackle hypothyroidism, which are much needed, as no current obesity treatment works for everyone.
Authors
Jessica Rampy, Alejandra Paola Torres-Manzo, Kendra Hoffsmith, Matthew A. Loberg, Quanhu Sheng, Federico Salas-Lucia, Antonio C. Bianco, Rafael Arrojo e Drigo, Huiying Wang, Vivian L. Weiss, Nancy Carrasco
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) and steatohepatitis (MASH) are leading causes of cirrhosis and hepatocellular carcinoma. Defects in autophagy contribute to the development of MASLD, however, the role of the Unc-51-like autophagy-activating kinase 1 (ULK1) in the pathophysiology of MASLD remains unclear. Herein, we show that ULK1, a serine/threonine kinase and core autophagy protein, is significantly repressed in human MASH livers, and that hepatocyte-specific loss of ULK1, unexpectedly, promotes hepatic steatosis and progression to liver fibrosis, without affecting basal autophagy flux. Phospho-proteomics identified the transcriptional coactivator NCOA3 as a downstream phospho-target of ULK1. Mechanistically, ULK1 phosphorylates NCOA3 to repress its transcriptional activity and restrain the CREB/CBP-mediated de novo lipogenic program. Accordingly, a phosphorylation-deficient NCOA3 mutant drives CREB/CBP-mediated lipogenesis, whereas genetic or pharmacological NCOA3 inhibition prevents steatosis, hepatic inflammation, and profibrotic signaling. Hence, ULK1-mediated NCOA3 phosphorylation is a fundamental and druggable checkpoint against the entire MASLD spectrum.
Authors
Young Do Koo, Romilia Tatiana Castillo, Asha Sukumaran Nair, Michael Garneau, Chad Gochee, Zachary V. Campbell, Tashya Shreyas Vakil, Jua Ha, Alex Marti, Jamie Soto, Debajyoti Das, Nuria Martinez-Lopez, Shipra Sharma, Yennifer Delgado, Callie Phung, Immy A. Ashley, Edmund D. Kapelczak, Rashel Jacobo, Eric T. Weatherford, Dao-Fu Dai, Jihane N. Benhammou, Andrea G. Marshall, Antentor Hinton Jr, Ling Yang, Renata O. Pereira, Tara TeSlaa, Mehdi Bouhaddou, Rajat Singh, E. Dale Abel

Abstract
Acquired generalized lipodystrophy (AGL) is a rare metabolic disorder frequently associated with autoimmunity. Its etiology is incompletely understood, and the effect of adipose tissue loss on intestinal inflammation in AGL remains unclear. Using mass cytometry and single-cell RNA-seq, we observed an oligoclonal expansion of T cells in the periphery and inflamed intestine in a patient with AGL and Crohn’s disease (AGLCD). To explore if loss of adipose tissue triggers lymphoproliferation, we studied lipodystrophic mice as a model for AGL. Unexpectedly, lipodystrophic mice did not show T cell expansion, were protected from colitis, and displayed a defect in the development of proinflammatory T cells, which could be reversed by allogeneic fat transplantations, indicating that clonal T cell expansion in AGLCD is not primarily caused by lipodystrophy. Instead, gene sequencing revealed a T cell–intrinsic de novo neuroblastoma RAS viral oncogene homolog (NRAS) mutation, implicating somatic mosaicism as a facilitator of clonal T cell expansion and intestinal inflammation in AGLCD.
Authors
Marilena Letizia, Toka Omar, Patrick Weidner, Manuel O. Jakob, Inka Freise, Susanne M. Krug, Britt-Sabina Löscher, Elisa Rosati, Benedikt Obermayer, Reyes Gamez-Belmonte, Julia Hecker, Jörn-Felix Ziegler, Benjamin Weixler, Patrick Asbach, Desiree Kunkel, Michael Stumvoll, Konstanze Miehle, Christoph Becker, Christoph S.N. Klose, Rainer Glauben, Dieter Beule, Anja A. Kühl, Thomas Conrad, Frank Tacke, Stefan Wirtz, Andre Franke, Ashley D. Sanders, Britta Siegmund, Carl Weidinger








Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)