Immunology
Abstract
The dynamic assembly and regulation of the IκB kinase (IKK) complex in the NF-κB pathway are central to the pathogenesis and progression of inflammatory bowel disease (IBD). We recently reported that the transcription factor hematopoietically-expressed homeobox (HHEX) promotes colitis-associated colorectal cancer, but the potential role of HHEX in intestinal inflammation remains uncharacterized. Here, we found that HHEX is upregulated in inflamed colons in a colitis mouse model and in clinical IBD samples. HHEX overexpression increased inflammatory cytokine expression, and HHEX loss largely abrogated the inflammatory response in vitro and intestinal inflammation in vivo. Mechanistically, IKKα phosphorylates HHEX at S213 to stabilize HHEX in response to TNF-α by inhibiting the interaction of HHEX with the E3 ubiquitin ligase MID2 and subsequent K48-linked ubiquitination and protein degradation. Importantly, HHEX interacts with and stabilizes the IKKα/IKKβ complex via its N-terminal domain, thereby activating the NF-κB pathway and establishing a positive feedback loop that exacerbates intestinal inflammation. Our study reveals a transcription-independent function of HHEX in promoting IKK complex assembly and colitis, identifying HHEX as an IBD susceptibility gene and a potential target for IBD treatment.
Authors
Zhebin Hua, Weimin Xu, Wenjun Ding, Zhuoyue Fu, Yaosheng Wang, Yiqing Yang, Fangyuan Liu, Zhujiang Dai, Wenbo Tang, Weijun Ou, Wensong Ge, YingWei Chen, Zhongchuan Wang, Chen-Ying Liu, Peng Du

Abstract
V-domain immunoglobulin suppressor of T cell activation (VISTA) is an immune checkpoint protein that impairs antitumor T cell responses. While broadly expressed on myeloid cells and T cells, the specific contribution of T cell–intrinsic VISTA to antitumor immunity remains undefined. This study investigated the phenotypic and functional consequences of T cell–specific VISTA deletion in tumor-specific CD8+ T cells. Single-cell transcriptomic analysis, TCR repertoire profiling, and flow cytometry revealed that loss of T cell–intrinsic VISTA enhanced early priming and short-term expansion of CD8+ T cells, yet this initial advantage failed to confer durable tumor control. Persistent dysfunction in VISTA-deficient T cells was in part driven by trans-VISTA on myeloid cells, while CTLA-4 upregulation further constrained T cell responses. T cell–intrinsic VISTA deficiency cooperated with CTLA-4 blockade to improve T cell survival and broaden TCR repertoire diversity, resulting in more robust tumor regression than CTLA-4 inhibition alone. A transcriptional signature enriched in VISTA-deficient cytotoxic T cells correlated with favorable outcomes in cancer patients treated with existing immune checkpoint inhibitors. These findings collectively define T cell–intrinsic mechanisms by which VISTA enforces T cell dysfunction and underscore its potential as both a therapeutic target and a biomarker of resistance to current immunotherapies.
Authors
Cassandra Gilmour, Elizabeth DeLaney, Prerana B. Parthasarathy, Dia Roy, Hieu M. Ta, Amin Zakeri, Paolo Elguera Grandez, Sachin Patnaik, Keman Zhang, Ivan Juric, Rahul Rangan, Zahraa Al-Hilli, Anthony Tufaro, Booki Min, Samuel E. Weinberg, Timothy A. Chan, Natalie L. Silver, Stefanie Avril, Tyler Alban, Li Lily Wang

Abstract
BACKGROUND Sepsis encompasses considerable biological and clinical heterogeneity. Previously, 2 phenotypes (“hyperinflammatory” and “hypoinflammatory”) have been consistently identified within sepsis via latent class analysis. These phenotypes differ in their biological features, clinical outcomes, and therapeutic responses to interventions. Prior studies of sepsis heterogeneity have focused primarily on the host response. Here, we investigate the potential influence of the causative pathogen on sepsis heterogeneity and pathobiology.METHODS We performed a retrospective observational analysis of 8,280 critically ill patients with sepsis to identify associations between pathogen characteristics and the hyperinflammatory and hypoinflammatory patient phenotypes. We also performed controlled murine and swine modeling of sepsis and lung injury and a secondary analysis of 449 patients enrolled in the EUPHRATES randomized controlled trial.RESULTS Pathogen characteristics (pathogen identity, burden, virulence, and anatomic site of infection) were strongly and independently associated with the previously reported phenotypes. In a cohort of critically ill patients with sepsis, infection with gram-negative pathogens, primarily Enterobacterales spp. (e.g., Escherichia coli, Klebsiella pneumoniae), was strongly associated with the hyperinflammatory phenotype. The hyperinflammatory phenotype was also independently associated with increased pathogen burden, virulence, and initial anatomic site of infection. In controlled murine and swine modeling, both the identity and burden of the pathogen provoked key biological features of the hyperinflammatory phenotype. Among patients with sepsis, the prognostic value of lactate clearance varied substantially by phenotype. In a secondary analysis of a randomized trial of polymyxin B hemoadsorption (which removes circulating endotoxin), hypoinflammatory patients experienced worse survival.CONCLUSIONS Our results demonstrate the central importance of pathogen features in the clinical and biological heterogeneity of sepsis. Future studies of sepsis pathobiology and heterogeneity should expand their scope beyond the host response, as understanding pathogen-host interactions will be crucial in the development of precision therapeutic strategies to improve patient outcomes.TRIAL REGISTRATION EUPHRATES trial NCT01046669.FUNDING 5P30AG024824, IK2CX002766, R01HL144599, K24HL159247, R01HL158626, R01HL173531, R35GM142992, R35GM145330, R35GM136312, K23HL166880, R35HL140026.
Authors
Rishi Chanderraj, Brian Bartek, Kathleen A. Stringer, Mohamad H. Tiba, Michael W. Sjoding, Ying He, Mark Nuppnau, Kale S. Bongers, Mark D. Adame, Sunny S. Lou, V. Eric Kerschberger, Matthew M. Churpek, Carolyn S. Calfee, Sandhya Tripathi, Debra M. Foster, John A. Kellum, Robert P. Dickson, Pratik Sinha

Abstract
Repetitive injuries are an important trigger of progressive fibrosis. To study if repetitive injuries induce an accelerated profibrotic process, also called “fibrosis-memory,” we established an experimental system with two consecutive, clearly separated insults in a model of renal fibrosis with reversible and irreversible unilateral ureteral obstruction. We found that a preceding fibrotic event of one kidney markedly enhanced subsequent development of fibrosis in the contralateral kidney. Aggravation of fibrosis during the second insult was dependent on memory CD4+ T cells. T cell depletion abrogated the fibrosis-memory effect, while adoptive transfer of memory T cells from fibrotic mice enhanced fibrosis in the recipients. Moreover, IL-3 production by memory CD4+ T cells was essential for aggravation of fibrosis in memory situations. In patients with systemic sclerosis, IL-3 expression by T cells was markedly increased, especially after a long disease duration accompanied by involvement of internal organs. In summary, our data identify IL-3–mediated fibrosis-memory as an important driver of progressive fibrosis.
Authors
Simone Buchtler, Antje Frühauf, Sophia Neumayer, Kathrin Schmidbauer, Yvonne Talke, Frederike Winter-Köhler, Saidou Balam, Karin Landgraf, Claudia Gebhard, Michael Rehli, Florian Volker Schlieckau, Maria Beck, Florian Günther, Martin Fleck, Kerstin Renner, Matthias Mack
Abstract
Immune evasion is a major obstacle ahead of pancreatic cancer therapy. Recent data implicate pro-inflammatory macrophages in the progression of pancreatic ductal adenocarcinoma (PDAC) and its therapeutic response. However, whether or which of the pro-inflammatory macrophage subtypes play a crucial role in the immune escape of PDAC remains unclear. Here we identify a population of CD138+ tumor-associated macrophages (TAMs), characterized by their pro-inflammatory and neutrophil-chemotactic activity, which undergo significant expansion in both PDAC patients and mouse models. These cells are elicited by a local synergy between IL-34-syndecan-1 and PGE2-EP2 signaling and are associated with immune evasion and poor clinical outcomes in patients, while also promoting immune escape and disease progression in mouse models. Mechanistically, CD138+ TAMs establish a feedforward loop with immunosuppressive Siglec-F+ neutrophils, which exhibit elevated PGE2 expression, via the secretion of Saa3 and Cxcl1. Targeting CD138+ TAMs by disrupting IL-34-syndecan-1 signaling with anti-IL-34 neutralizing antibodies significantly suppresses PDAC progression, especially when combined with anti-PD-1 antibodies. Together, our study elucidates a CD138+ TAM-Siglec-F+ neutrophil axis that drives immune escape in PDAC and proposes a therapeutic strategy that integrates IL-34-syndecan-1 signaling blockade with anti-PD-1 immunotherapy for the treatment of PDAC.
Authors
Chao Wang, Qi Zhang, Jinyan Huang, Fangyu Lin, Danyang Zhao, Youling Mu, Junshuo Tong, Jinping Li, Yingjiqiong Liang, Tao Zeng, Fukang Shi, Hang Shen, Tingting Lu, Tingbo Liang
Abstract
BACKGROUND. Immune checkpoint inhibitors (ICIs) targeting the programmed cell death-1 axis have revolutionized metastatic non–small cell lung cancer (mNSCLC) treatment. However, disease progression remains a concern, and the role of the complex tumor microenvironment (TME) in treatment failure is not fully understood. METHODS. In this biomarker study involving 103 patients with mNSCLC—including 81 patients who received ICI treatment—we evaluated the association between heterogeneous immune cell subsets and ICI efficacy through single-cell spatial profiling of pretreatment tumor tissue, using a 29-marker multiplex immunohistochemistry platform built for in-depth dissection of the TME. RESULTS. Among various types of intratumoral lymphocytes including T-helper 1 cells, regulatory T cells, and natural killer cells, only CD8+ T cells (TILs) were associated with ICI efficacy. Computational tissue segmentation underscored the importance of direct physical interactions between CD8+ TILs and cancer cells for ICI efficacy. TIL phenotyping identified CD39/CD103/Ki-67 positivity as a hallmark of exhausted yet functional tumor-reactive CD8+ TILs. Immunosuppressive tumor-associated macrophages (TAMs) and cancer-associated fibroblasts were independent unfavorable adversaries. High CD73 expression on cancer cells was suggested to confer tolerance to ICI in EGFR/ALK-oncogene+ NSCLC, potentially through M2-TAM accumulation and aberrant angiogenesis. CONCLUSION. Our study delineates the clinical relevance of heterogeneous immune cell subsets in ICI-treated mNSCLC, aiding the development of targeted therapeutic strategies. TRIAL REGISTRATION. Not applicable because this is a retrospective study. FUNDING. Osaka Cancer Society, KANAE Foundation for the Promotion of Medical Science, SGH Foundation, and YOKOYAMA Foundation for Clinical Pharmacology.
Authors
Kohsuke Isomoto, Koji Haratani, Takahiro Tsujikawa, Shuta Tomida, Yusuke Makutani, Masayuki Takeda, Kimio Yonesaka, Kaoru Tanaka, Tsutomu Iwasa, Kazuko Sakai, Kazuto Nishio, Akihiko Ito, Kazuhiko Nakagawa, Hidetoshi Hayashi
Abstract
Poly (ADP-ribose) polymerase inhibitors (PARPi) benefit homologous recombination-deficient (HRD) malignancies, yet resistance remains a major challenge. Leveraging specimens from a prospective neoadjuvant niraparib monotherapy trial in treatment-naïve high-grade serous ovarian cancer, we integrated PhenoCycler-Fusion spatial profiling, scRNA-seq, and multiplex immunohistochemistry to identify two therapeutic-modulated cellular neighborhoods: an IFN+ tumor cell-enriched niche that expands in resistant lesions and a tumor-associated macrophage (TAM)-enriched niche that persists but acquires enhanced immunosuppressive features. Mechanistically, sustained tumor cell-derived IFN induced osteopontin (SPP1) expression in TAMs via STAT signaling, creating immunosuppressive niches enriched in Tregs and myofibroblastic cancer-associated fibroblasts with intensified cell-cell interactions. SPP1 directly suppressed T cell signaling and effector function. High baseline SPP1+ cells predicted lower response rate (30.0% vs 76.2%, P = 0.021) and shorter progression-free survival (median 13.5 vs 28.3 months, P = 0.0006). In HRD mouse models, SPP1 blockade restored PARPi sensitivity, reversed acquired resistance, and enhanced T cell cytotoxicity—effects abrogated in immunodeficient mice, confirming immune dependence. These data establish a spatial IFN–SPP1 axis whereby persistent tumor cell IFN reprograms TAMs to promote PARPi resistance, position SPP1 as a key therapeutic target and prognostic biomarker for this therapy, and underscore therapeutic potential of microenvironment-targeted strategies to overcome PARPi resistance.
Authors
Dan Liu, Kangjia Tao, Cheng Xu, Wen Yang, Chujun Cai, Cui Feng, Kairong Xiong, Sisi Wu, Yaying Lin, Zikun Peng, Jianhua Chi, Wen Pan, Qing Zhong, Jiahao Liu, Xiong Li, Xingzhe Liu, Dongchen Zhou, Ding Ma, Guang-Nian Zhao, Yu Xia, Yong Fang, Qinglei Gao
Abstract
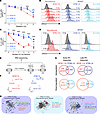
Patients with malignant peripheral nerve sheath tumors (MPNSTs) have poor outcomes despite multimodal treatment with surgery, radiation, and systemic therapy. The responses to radiotherapy (RT) are mixed, and the biologic mechanisms underlying this heterogeneity in the radiation response of MPNSTs are not understood. Here, we combined bulk and single-cell transcriptomics, genome-wide CRISPR interference screens, and multiplatform molecular analysis across MPNST cells, mouse allograft models, and patients’ samples to understand the mediators of the radiation response. Our data revealed that MPNSTs, but not benign plexiform neurofibromas, induced a type I IFN signature that functionally mediated the radiation response. Moreover, irradiation of immunocompetent mouse MPNST allografts led to IFN-mediated T cell recruitment and activation. Both host mouse T cells and intact tumor IFN receptor signaling were required for RT’s efficacy in mouse MPNST allografts. Analysis of human MPNST resection specimens demonstrated that increased microenvironmental and CD8+ T cell infiltration were associated with improved local control following RT. These results provide a preclinical rationale for combining immunomodulatory agents targeting IFN signaling to improve radiation responses in MPNSTs and potentially other soft tissue sarcomas.
Authors
Iowis Zhu, Julian Chien, Gabriel E. Rech, Kanish Mirchia, Sixuan Pan, Kaeli Miller, Joanna Pak, Rosanna Wustrack, Varun Monga, Steve E. Braunstein, Mark D. Adams, Line Jacques, Melike Pekmezci, S. John Liu, Harish N. Vasudevan

Abstract
Liver invasion is one of the most frequent events in the progression of gallbladder cancer (GBC). However, the cellular and pathological role of the tumor-liver–interface microenvironment in liver invasion is still enigmatic. Here, we applied single-cell and spatial transcriptomics to systematically investigate the cellular component and gene expression regulation of the microenvironment from the tumor to the liver, specifically the invasive boundary. Our analyses revealed that CXCL9+ macrophage–rich immune cell niches were accumulated in the tumor-liver invasive margin, where 2 subclasses of the CXCL9+ immune cell niches, CXCL9+TRAC+ (CT) and CXCL9+C1QB+ (CC) niches, were identified. CD8+ T cells were recruited by CXCL9+ macrophages through CXCL9-CXCR3 interaction in the CT niche, which was located adjacent to the liver. Moreover, the CC niche was proximal to the tumor core, where tumor cells induced CD8+ T cell exhaustion via LGALS4 expression. In addition, our cohort study showed that high CXCL9 and low LGALS4 in the liver invasion margin demonstrated a favorable prognosis and better responses to anti–PD-1 immunotherapy for patients with gallbladder cancer. Altogether, these findings demonstrate novel cellular and molecular mechanisms underlying liver invasion and offer clinical value for immunotherapies.
Authors
Maolan Li, Zhaonan Liu, Shenbing Shan, Ziyao Jia, Yongsheng Li, Fatao Liu, Lina Lu, Shimei Qiu, Chen Li, Ziyi Wang, Siyuan Yan, Yuhao Zhao, Lili Gao, Zhiqing Yuan, Yuanding Liu, Jiyao Ma, Jiayi Feng, Pengxiao Geng, Yiming Li, Xiaojing Xu, Xinhua Lin, Changjun Liu, Zebing Liu, Wenguang Wu, Xiangsong Wu, Wei Gong, Yanjing Li, Dongxi Xiang, Yongning He, Yun Liu, Rong Shao, Kwan Man, Wu Wei, Yingbin Liu
Abstract
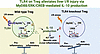
Surgical stress, such as hepatic ischemia-reperfusion (I/R) injury, induces excessive inflammation and adversely affects liver surgery outcomes. Regulatory T cells (Tregs) are crucial for immune homeostasis, yet their protective mechanisms against liver I/R injury remain unclear. In this study, we demonstrated that decreased hepatic Treg abundance correlates with increased liver injury in patients undergoing hepatic hemangioma resections. In murine models, Treg depletion worsened liver I/R injury. Bulk RNA-seq of hepatic Tregs showed enrichment of Toll-like receptor (TLR) signaling pathways, with flow cytometry identifying TLR4 as the most increased TLR after I/R. Treg-specific Tlr4 knockout mice (Treg-Tlr4–/– mice) exhibited exacerbated liver injury following I/R. Adoptive transfer of WT Tregs, but not Tlr4-deficient Tregs, alleviated liver injury in both Treg-depleted and Treg-Tlr4–/– mice. Transcriptomic analysis revealed that IL-10 production was impaired in Tlr4-deficient Tregs. Mechanistically, Tlr4-deficient Tregs showed reduced activation of the MyD88/ERK/CREB pathway, resulting in diminished IL-10 production. Myd88–/– and IL-10–/– Tregs failed to confer protection against liver I/R injury, whereas exogenous IL-10 administration rescued the hepatic dysfunction in Treg-Tlr4–/– mice. Our findings implicate the vital role of TLR4 in Tregs to mitigate liver I/R injury and offer a potential therapeutic option to reduce postoperative complications following liver surgery.
Authors
Hongji Zhang, Yunwei Zhang, Tianxing Ren, Carolyn Tsung, Peng Song, Peng Xu, Guoliang Wang, Chunyan Cao, Changyan Wang, Ping Sun, Qi Zhang, Yanhong Zhu, Xin Zhong, Yong Guan, Xiaofei Zhang, Han Wang, Jinxiang Zhang, Hui Wang






Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)