Advertisement
Clinical Research and Public HealthAIDS/HIVInfectious diseaseVirology
Open Access | ![]() 10.1172/JCI194081
10.1172/JCI194081
Factors associated with resistance of HIV-1 reservoir viruses to neutralization by autologous IgG antibodies
Natalie F. McMyn,1 Joseph Varriale,1 Hanna W. S. Wu,1 Vivek Hariharan,1 Milica Moskovljevic,1 Toong Seng Tan,2,3 Jun Lai,1 Anushka Singhal,1 Kenneth Lynn,4,5 Karam Mounzer,6 Pablo Tebas,5 Luis J. Montaner,4 Rebecca Hoh,7 Xu G. Yu,2,3 Mathias Lichterfeld,2,3 Francesco R. Simonetti,1 Colin Kovacs,8 Steven G. Deeks,7 Janet M. Siliciano,1 and Robert F. Siliciano1,9
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
McMyn, N.
in:
PubMed
|
Google Scholar
|

1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Varriale, J.
in:
PubMed
|
Google Scholar
|
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Wu, H. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Hariharan, V. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Moskovljevic, M. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Tan, T. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Lai, J. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Singhal, A. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Lynn, K. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Mounzer, K. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Tebas, P.
in:
PubMed
|
Google Scholar
|
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Montaner, L.
in:
PubMed
|
Google Scholar
|
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Hoh, R.
in:
PubMed
|
Google Scholar
|
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Yu, X. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Lichterfeld, M. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Simonetti, F.
in:
PubMed
|
Google Scholar
|
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Kovacs, C.
in:
PubMed
|
Google Scholar
|
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Deeks, S.
in:
PubMed
|
Google Scholar
|
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by Siliciano, J. in: PubMed | Google Scholar
1Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
2Brigham and Women’s Hospital, Boston, Massachusetts, USA.
3Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA.
4The Wistar Institute, Philadelphia, Pennsylvania, USA.
5University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Philadelphia Field Initiating Group for HIV-1 Trials, Philadelphia, Pennsylvania, USA.
7University of California San Francisco, San Francisco, California, USA.
8Maple Leaf Medical Clinic, Toronto, Ontario, Canada.
9Howard Hughes Medical Institute, Baltimore, Maryland, USA.
Address correspondence to: Robert Siliciano, Department of Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore Maryland, 21205, USA. Phone: 410.955.2958; Email: rsiliciano@jhmi.edu.
Find articles by
Siliciano, R.
in:
PubMed
|
Google Scholar
|
Published July 29, 2025 - More info
J Clin Invest. 2025;135(19):e194081. https://doi.org/10.1172/JCI194081.
© 2025 McMyn et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: April 2, 2025; Accepted: July 23, 2025
Related article:
Abstract
Initial efforts to control HIV infection include an autologous neutralizing antibody (aNAb) response. aNAbs bind Env trimers of the infecting HIV strain to neutralize virus but are not very effective at controlling HIV, as the virus quickly develops escape mutations to evade neutralization. Nevertheless, recent evidence suggests that aNAbs exert ongoing immune pressure on viral isolates in people living with HIV (PWH) treated with anti-retroviral therapy (ART) during chronic and early infection. In this issue of the JCI, McMyn et al. studied the dynamics of aNAb resistance in a cohort of 31 PWH treated with ART. Notably, a large proportion of HIV reservoir viral isolates were resistant to aNAb neutralization, which correlated with longer duration on uninterrupted ART, suggesting that selection for aNAb-resistant isolates occurs as reservoir cells containing neutralization-sensitive isolates are eliminated. aNAb resistance was not attributed to waning antibody response, which persisted for over 20 years despite viral suppression.
Authors
Nancie M. Archin
-
Results
Study participants. We studied 31 PWH, including 9 participants from our initial aNAb study (30) who had extensive reservoir sequencing. The mean time on combination ART was 15.9 years (range 4.5–26.7 years, Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI194081DS1). The demographics were 87.1% male, 12.9% female, 64.5% Black, 32.3%, White, and 3.2% Pacific Islander. Participants had a mean CD4 nadir of 225 cells/μL and a mean age of 54 years at the last sample collection. Viral outgrowth results for 25 participants were previously reported in Bertagnolli et al. (30) and McMyn et al. (15). Those studies showed that replication-competent virus could be readily isolated from study participants, even those on ART for more than 20 years, at frequencies between 0.05–16.25 infectious units per million (IUPM) resting CD4+ T cells, and that aNAbs could suppress outgrowth of a subset of these viruses.
All participants initiated ART during chronic infection. Longitudinal plasma HIV-1 RNA levels and CD4 counts are in Supplemental Figure 1 and McMyn et al. (15). Five participants had short periods of ART interruption. Three of these (DEL-SPC-015, -017, and -019) were in ACTG clinical trial A5340 (IDs A02, A06, and A13, respectively) which involved administration of the bNAb VRC01 and an analytic treatment interruption (ATI). Plasma HIV-1 RNA levels during the ATI were previously reported (54). DEL-SPC-012 and JH448 had treatment interruptions due to nonadherence. All participants had plasma HIV-1 RNA levels below the detection limit (< 20–40 copies/mL) at the time of sampling. Small, isolated blips, transient increases in HIV-RNA above the detection limit (75, 76), were not considered in calculating time on uninterrupted ART.
Sensitivity of reservoir isolates to aNAbs. To determine the aNAb sensitivity of replication-competent proviruses from a diverse group of PWH, we performed quantitative viral outgrowth assays (QVOAs) (77, 78) using resting CD4+ T cells. Cultures from 28 of 31 participants had greater than or equal to 5 p24+ outgrowth wells, for a total of 591 independent isolates. Full length env sequencing revealed 138 different (distinct) env sequences. The other 453 sequences were identical to other sequences from the same participant. Among sequences found more than once, there were 69 sets of identical sequences ranging in size from 2 to 59 isolates. Distinct env sequences were cloned into expression vectors for pseudovirus generation and neutralization assays as previously described (30). Because nonspecific inhibition can be observed at IgG concentrations over 100 μg/mL, we arbitrarily designated isolates as resistant if the IC50 for autologous IgG was over 100 μg/mL.
Of 138 distinct HIV-1 env pseudoviruses from 28 PWH 55% (76/138) were resistant to neutralization (Figure 1A). Since some individuals had fewer distinct isolates to test due to high reservoir clonality and/or smaller reservoir size, we determined the fraction of resistant isolates per PWH to normalize for different numbers of isolates per participant (Figure 1B). There was wide variation in the fraction of resistant sequences (0%–100%, median 80%).
 Figure 1
Figure 1Variation in sensitivity of reservoir isolates to aNAbs. (A) aNAb IC50 values were determined in TZM.bl-based neutralization assays for distinct pseudoviruses generated from outgrowth viruses from PWH on ART (n = 138 isolates). Autologous IgG concentrations up to 100 μg/mL were used. Circles represent distinct isolates, and colors represent participants. (B) Percentage of distinct outgrowth viruses resistant (IC50 > 100 μg/mL) to neutralization by contemporaneous aNAbs per PWH. Each data point (n = 28) represents the % of resistant viruses among the total number of viruses tested in each PWH. Bar represents median. (C) aNAb neutralization of distinct isolates shown in A with additional data points representing independent isolates from the same PWH with env sequences identical to those shown in A, n = 591. IC50 values determined for one member of a set of isolates with identical env sequence were used for all members of the set. (D) Percentage of outgrowth viruses resistant to neutralization (IC50 > 100 μg/mL) by aNAbs per PWH, n = 28, using values from (C). All isolates from each participant, including sequence-identical isolates, are considered in the percentage calculation. Bar represents median.
Effects of reservoir clonality. The analyses presented above consider only distinct variants. However, reservoir composition is also influenced by clonal expansion. Therefore, we analyzed resistance to aNAbs considering all isolates from each donor, not just distinct sequences. Of 591 total isolates, 60% (357/591) were resistant to contemporaneous aNAbs (IC50 > 100 μg/mL, Figure 1C). After normalizing for different numbers of isolates for each participant, we found a median of 92% resistant viruses per PWH, again with very high person-to-person variation (0%–100%) (Figure 1D). For 43% of participants, all reservoir isolates were resistant (Figure 1, B and D).
Factors associated with aNAb resistance. Figure 1D reveals a separation in aNAb resistance between 20 participants with high resistance (67%–100% resistant isolates per PWH) and 8 participants with high sensitivity (0%–26% resistant isolates per PWH). Of 74 distinct viral isolates from the high resistance group, 64 isolates (86%) were neutralization resistant (Figure 2A). This fraction was significantly higher than the 19% observed in the group with high aNAb sensitivity (12/64 distinct aNAb-resistant isolates, P < 0.0001, Figure 2A). The difference was visually apparent in flatter neutralization curves for the aNAb-resistant group (Figure 2, B and C). The impact of dose-response curve slope is described below. For both groups, resistant variants were found among sets of isolates with identical env sequences as well as sequences observed only once (Supplemental Figures 2 and 3). There were no correlations between aNAb IC50 values and the number of identical sequences in a set for either group (Supplemental Figure 4).
 Figure 2
Figure 2Understanding individual variation in aNAb resistance. PWH on ART were divided into 2 groups, those with aNAb-resistant or aNAb-sensitive reservoirs, based on Figure 1D. The aNAb-resistant group had greater than or equal to 67% resistant isolates while the aNAb-sensitive group had less than or equal to 26% resistant isolates. (A) aNAb IC50 values for distinct pseudoviruses for the aNAb-resistant (n = 74 isolates) and aNAb-sensitive groups (n = 64 isolates). Circles represent distinct isolates, and colors represent participants. The Mann-Whitney test was used to calculate significance. ****P < 0.0001. Bar represents median. (B and C) Dose-response curves for the inhibition of pseudovirus infectivity by autologous IgG for isolates from the aNAb-resistant (B, n = 74 isolates) and aNAb-sensitive (C, n = 64 isolates) groups. The median curve is overlayed in black. Data are reported as a mean % of maximum infectivity from triplicate measurements. (D) Percentage of resistant viruses per PWH from Figure 1B correlates with time on uninterrupted ART (Spearman’s correlation, n = 28). (E) Percentage of resistant viruses per PWH, including sets of identical sequences from Figure 1D, correlates with time on uninterrupted ART (Spearman’s correlation, n = 28).
We next explored other explanations for differences in aNAb resistance. No significant differences between the 2 groups were found for CD4 nadir, a proxy for time of untreated infection (Supplemental Figure 5A), or reservoir size, based on QVOA measurements (Supplemental Figure 5B). We then evaluated differences in total time on ART. Although the mean time on ART for the aNAb-resistant group was higher (16.9 vs 10.9 years), the difference was not significant (P = 0.1018, Supplemental Figure 5C). This time included the time that 5 participants (DEL-SPC-012, -015, -017, -019, and JH448) experienced treatment interruptions. Therefore, we compared time on uninterrupted ART or time since last period of measurable viremia. This analysis resulted in significant differences. For the aNAb-resistant group, the average time of uninterrupted ART was 16.5 years, significantly higher than the average time of 8.2 years for the aNAb-sensitive group (P = 0.0277, Supplemental Figure 5D). These results suggest that ART interruptions and shorter times on uninterrupted ART may affect neutralization sensitivity.
To further investigate the relationship between aNAb resistance and time on ART, we analyzed the correlation between aNAb IC50 values or % resistance per person and time on ART or time on uninterrupted ART. There was a significant positive correlation between the aNAb IC50 values of distinct isolates from both the aNAb-sensitive and aNAb-resistant groups and time on ART (Spearman r = 0.2058, P = < 0.0155; Supplemental Figure 6A). We found a more significant positive correlation between aNAb IC50 values of distinct isolates and time on uninterrupted ART (Spearman r = 0.4644, P = < 0.0001, Supplemental Figure 6B). When the analysis was expanded to include all independent isolates from each PWH and not simply the distinct isolates, the correlation was highly significant for both time on ART (Spearman r = 0.2441, P = < 0.0001) and time on uninterrupted ART (Spearman r = 0.3909, P = < 0.0001). Because these analyses are affected by the number of isolates per donor, we also examined the correlation between the fraction of distinct isolates that were resistant in each PWH and the time on uninterrupted ART. We found a significant positive correlation (Spearman r = 0.5154, P = 0.0050; Figure 2D). When the analysis included all isolates from each PWH, including identical sequences, there was a weaker positive correlation (Spearman r = 0.4327, P = 0.0215, Figure 2E). When the total time on ART was used, no significant correlation was found. Although conclusions depended on how time on ART was defined, longer times on ART were generally associated with greater aNAb resistance, perhaps indicating gradual selection against cells carrying aNAb-sensitive viruses (see below).
To determine whether the observed aNAb resistance of replication-competent isolates was representative of other persistent proviruses not detected in outgrowth assays, we assessed aNAb sensitivity of intact env sequences amplified from resting CD4+ T cells from 6 PWH on long-term ART (> 21 years) (15). The high aNAb resistance of outgrowth viruses was also observed for these proviruses (Supplemental Figure 2). Across the 6 participants, 16 of 17 distinct proviruses were resistant. The 1 sensitive isolate had an IC50 of 96.12 μg/mL. These results provide evidence for a latent reservoir dominated by resistant variants regardless of inducibility. The finding that most reservoir viruses are resistant to neutralization by contemporaneous IgG, especially in PWH on long-term ART, suggests that, over long time intervals, there may be a selection against cells carrying aNAb-sensitive viruses. Alternatively, a decline in aNAb concentration may explain reduced neutralization (see below).
aNAb resistance at physiologic IgG concentrations. The above results indicate that in many PWH on ART, most replication-competent reservoir viruses are not strongly neutralized by contemporaneous autologous IgG in in vitro assays. However, these assays are carried out using polyclonal IgG concentrations in the μg/mL range, while the in vivo plasma concentration of IgG is 7–16 mg/mL (79). From conventional dose-response curves, it is difficult to ascertain the degree of inhibition at in vivo concentrations (Figure 3A). Therefore, we used previously described pharmacodynamic metrics to predict the in vivo effects of aNAbs (80–82). The dose response curve can be described using the median effect equation (83):
 Figure 3
Figure 3Inhibitory effect of aNAbs at physiologic IgG concentrations. (A) Hypothetical dose-response curves for inhibition of infection by autologous IgG antibodies. The fraction of infection events not inhibited (fraction unaffected or fu) declines with increasing antibody concentrations. Curves show inhibition by 3 different antibody preparations that have the same IC50 (1 μg/mL, dotted line) but different slopes (m), a measure of cooperativity. In vitro neutralization assays are conducted at IgG concentrations well below physiologic IgG concentrations (shaded area). (B) Curves from A plotted with a logarithmic y-axis, illustrating the dramatic effect of m on inhibition. (C) Instantaneous inhibitory potential (IIP), the logs of inhibition of a single round of infection, for the same antibody preparations. (D) Effect of m on IIP for distinct isolates from the aNAb-resistant (orange, n = 31) and aNAb-sensitive (blue, n = 48) groups. IC50 value trend lines are shown at 3 indicated concentrations. For many resistant isolates, m, IC50, and IIP, could not be determined because infection did not consistently decrease with increasing IgG concentrations. (E) Distribution of slope values for the isolates from D. Circles represent distinct isolates, and colors represent participants. Bar represents median. Significance was calculated using the Mann Whitney test. **P < 0.01. (F) Distribution of IIP values for the isolates from D. Dotted line indicates the IIP value above which effective suppression of viral replication by combination ART regimens is observed (85). IIP values for single antiretroviral drugs at average plasma concentrations are shown (80, 85). Significance was calculated using the Mann Whitney test. ****P < 0.0001.
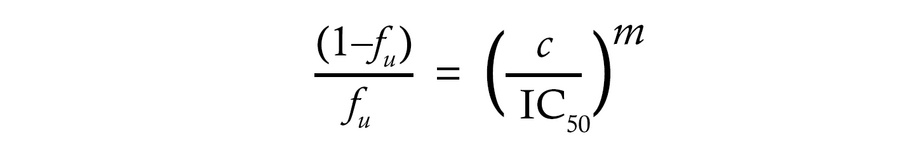
Equation 1
where ƒu is the fraction of infection events unaffected (not inhibited) by antibodies at concentration (c) given the IC50 and the dose response curve slope (m) or Hill coefficient, a measure of cooperativity that is equal to 1 for noncooperative processes. Slopes greater than 1 give steeper dose-response curves, but differences in inhibition at physiologic IgG concentrations are only apparent if the y-axis values are displayed on a log scale (Figure 3B). The inverse of this plot gives the instantaneous inhibitory potential (IIP),
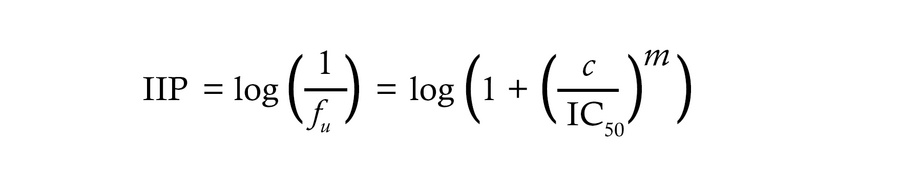
Equation 2
an intuitive metric of antiviral activity, which is the number of logs by which single round infection events are reduced at a given antibody concentration (Figure 3C) (80–82). IIP depends on both IC50 and m (Figure 3D). Because of their exponential relationship, IIP is strongly influenced by m. Figure 3D shows calculated IIP at 10 mg/mL of IgG based on experimental measurements of IC50 and m for distinct isolates from the aNAb-resistant and aNAb-sensitive groups. As expected, IIP values are strongly influenced by m (linear regression R2 = 0.77 and 0.83 for isolates from the resistant and sensitive groups, respectively). Figure 3E shows slope values for the 2 groups. Although antigenic and antibody heterogeneity can reduce slope, we observed slopes greater than 1, indicative of positive cooperativity, as is seen for HIV-1 protease inhibitors, nonnucleoside reverse transcriptase inhibitors, entry inhibitors, and some bNAbs (80–82, 84).
This analysis allows extrapolation of dose-response curves to physiologic IgG concentrations (Figure 3F). For the aNAb-resistant group, most isolates were weakly inhibited with a median IIP of 1.3, just above the level of nonspecific inhibition observed with control IgG. For the aNAb-sensitive group, many isolates were inhibited with a median IIP of 2.4, meaning greater than 2 logs of inhibition at in vivo IgG concentrations, comparable to single antiretroviral drugs (80–82). Effective combination ART regimens produce greater than 5 logs of inhibition (85). Only 4 isolates were inhibited by aNAbs with IIP greater than 5 (Figure 3F). These isolates had IC50 values less than 13 μg/mL and slopes greater than 1.7 (Supplemental Table 2). This analysis excluded 43 distinct isolates from the aNAb-resistant group and 16 from the aNAb-sensitive group because slopes and IIP values could not be calculated due to poor inhibition (nondecreasing dose response curves, maximum inhibition < 20%, or median effect plot R2 < 0.8). Together, these results indicate that, like single antiretroviral drugs, aNAbs can reduce replication of many reservoir viruses but not sufficiently enough to prevent selection of resistant variants.
Sensitivity of reservoir viruses to neutralization by bNAbs. In light of the high proportion of aNAb-resistance after long-term ART, we explored whether these viruses were resistant to 3 clinically relevant bNAbs: VRC01, 10-1074, and PGDM1400, which bind to the CD4 binding site, the V3 glycan site, and the V2 apex, respectively (45). Many isolates from the aNAb-resistant group were neutralized by bNAbs (median bNAb IC50: 1.280, 2.857, and 1.595 μg/mL for VRC01, 10-1074, and PGDM1400, respectively; Figure 4A). For 90% of donors in the aNAb-resistant group, all isolates were sensitive to at least one bNAb with IC50 values less than 4 μg/mL, and 55% of donors had isolates sensitive to at least 2 bNAbs (Supplemental Table 3). Thus, although viral isolates from these PWH were aNAb resistant, many isolates were sensitive to neutralization by 1 or more bNAbs. Only 4 isolates from 2 PWH were resistant to all 3 bNAbs. Importantly, bNAb sensitivity was not a predictor of aNAb sensitivity. The 10 isolates with aNAb IC50 values less than 100 μg/mL had similar bNAb sensitivities to isolates with aNAb IC50 values greater than 100 μg/mL (Supplemental Table 3). Ultimately, the majority of viruses in the aNAb-resistant group could be effectively neutralized by some bNAbs, ruling out general neutralization resistance as an explanation for high aNAb resistance in that group. Similar results were obtained for the aNAb sensitive group (Figure 4B).
 Figure 4
Figure 4Sensitivity of outgrowth viruses to bNAbs. (A) TZMbl-based neutralization assays were conducted with VRC01, 10-1074, and PGDM1400 and pseudoviruses generated from outgrowth viruses (n = 74) from the aNAb-resistant group. (B) Neutralization assays with pseudoviruses generated from outgrowth viruses (n = 58) from the aNAb-sensitive group. IC50 values above the highest antibody concentration tested are shown above the dotted lines at 4 μg/mL. Values below the lowest concentration tested are shown below the dotted lines at 0.032 μg/mL. Circles represent distinct isolates, and colors represent participants. We used a nonparametic 1-way ANOVA (Friedman test) followed by Dunn’s test for multiple comparisons to calculate significance. Bar represents median.
Some aNAb responses persist over long times on ART. Another explanation for the large proportion of outgrowth viruses resistant to aNAbs is that, without antigen, the anti-HIV antibody response wanes. Therefore, we studied antibody responses in participants on very long-term ART. Western blot analysis of IgG from a set of participants who had been on ART for an average of 22.6 years showed that some anti-HIV antibodies persisted despite prolonged suppression of viremia (Supplemental Figure 7). To examine whether neutralizing antibody responses persist, we first tested neutralization of pseudoviruses carrying the env gene of HIV-1 SF162, a tier 1 virus that is highly sensitive to neutralization by antibodies from many PWH (86, 87). Neutralization assays with SF162 pseudovirus were performed with IgG from 16 participants who had been on very long-term ART (mean 22.8 years), 12 of whom were from the aNAb-resistant group. SF162 pseudovirus was neutralized by IgG from 15 of 16 participants (Figure 5, A and B). The interquartile range of IC50 values was 6.9–26.4 μg/mL, with a median of 18.1 μg/mL (Figure 5B). No neutralizing activity was detected with IgG from an HIV-seronegative donor. For 3 participants with only 1 or no QVOA outgrowth viruses (SCOPE2006, JH167, JH24), participant IgG neutralized SF162 pseudovirus with IC50 values less than 26 μg/mL. This suggests that a smaller reservoir did not result in reduced antibody stability over time. Together, these results demonstrate that some neutralizing antibodies to the HIV-1 Env protein are detectable in most PWH even after more than 20 years of treatment, regardless of reservoir size.
 Figure 5
Figure 5Persistence of neutralizing antibodies to HIV-1 Env in PWH on long-term ART. (A) Dose-response curves for inhibition of SF162 pseudovirus infection of TZM.bl cells by IgG purified from 16 PWH on long-term ART and 1 uninfected donor. (B) IC50 values from dose-response curves in A. A value above the highest concentration tested (300 μg/mL) is shown in an open symbol. Dashed line represents median, and solid bars represent interquartile range. n = 16. (C) Dose-response curves for the inhibition of SF162 pseudovirus infection of TZM.bl by early and 20-year IgG samples from SCOPE2046, (D) SCOPE2256, and (E) SCOPE2114. (F) Dose-response curves for the inhibition of TZM.bl infectivity of one autologous outgrowth virus by early and 20-year autologous IgG collection timepoints from SCOPE2046, (G) SCOPE2256, and (H) SCOPE2114. Data for (A and C–H) are reported as percentage (mean, SEM) of infectivity from triplicate measurements.
Antibodies that neutralize SF162 recognize an epitope that is conserved and immunogenic. Thus, these antibodies are distinct from the aNAbs, which generally show limited heterologous neutralization (30). Examining the stability of aNAb responses in PWH on long-term ART is difficult because longitudinal plasma samples spanning 2 decades are rarely available. To determine if aNAb titers persist over long time intervals, we identified 3 study participants in the aNAb-resistant group who had been on ART for over 20 years and had a plasma sample from more than a decade earlier. First, we compared aNAb neutralization of SF162 between the early and 20-year IgG samples. For 2 participants, neutralizing activity remained similar, while in 1 participant there was a more than 1 log increase in IC50 (Figure 5, C–E).
We then compared neutralizing activity between the early and 20-year IgG preparations against autologous outgrowth viruses from the 20-year timepoint (Figure 5, F–H). For participant SCOPE2046, 11 of 11 isolates had identical env sequences, potentially reflecting an expanded clone (Supplemental Figure 2). For this Env, the aNAb IC50 increased from 81.44 to over 100 μg/mL over 20.9 years of ART, demonstrating some waning of the aNAb response (Figure 5F). Interestingly, neutralization of SF162 pseudovirus remained relatively stable over the same period (Figure 5C). For the second participant (SCOPE2256), 23 of 26 isolates had identical env sequences (Supplemental Figure 2). For this Env, modest neutralizing activity persisted over 13 years of ART (Figure 5G) as did neutralizing activity against SF162 (Figure 5D). The other 2 outgrowth isolates from SCOPE2256 were identical and remained resistant (IC50 > 100 μg/mL) at both timepoints (not shown). For the third participant (SCOPE2114), 1 of 5 distinct outgrowth viruses was neutralized by IgG from the late time point (IC50 = 86.41 μg/mL), while IgG from 18.5 years earlier had less neutralizing activity (IC50 > 100 μg/mL) (Figure 5H). An opposite trend was observed for neutralization of SF162 pseudovirus (Figure 5E). The other 4 outgrowth viruses from SCOPE2114 remained resistant (IC50 > 100 μg/mL) at both timepoints (not shown). Together, these observations demonstrate that aNAb responses can persist, improve, or wane, depending on the individual and the viral sequence tested. The waning of aNAb responses might explain the observed neutralization resistance in some PWH on ART, but in cases where the aNAb response remains stable, it is possible that the relevant antibodies eliminate reservoir cells carrying sensitive variants, leaving only aNAb-resistant viruses.
Autologous IgG antibodies can mediate ADCC against cells infected with reservoir viruses. We next explored whether the observed prevalence of aNAb-resistant viruses reflects a selection against cells with sensitive viruses through ADCC. Only a small subset of reservoir cells are transcriptionally active at any given time, and thus this differential killing would only become apparent over long time intervals. Therefore, we studied IgG-dependent, NK cell–mediated killing of target cells expressing autologous Envs from participants on ART for over 20 years. We generated replication-competent, NL4-3–based reporter viruses (NL4-3-ΔNef-eGFP) incorporating the env gene from participant-specific outgrowth viruses (Figure 6A). Next, we infected CEM.NKR.CCR5 cells (88), chosen to reduce nonspecific killing. The target cells were then incubated with NK cells purified from uninfected donors in the presence of participant IgG. Killing was measured as a reduction in live, GFP+ target cells (Supplemental Figure 8). We tested aNAb-sensitive and aNAb-resistant isolates from 3 long-term ART participants. Minimal nonspecific killing occurred in control cultures with uninfected donor IgG, in cultures without IgG, and in cultures without NK cells (Figure 6B). For both aNAb-sensitive and aNAb-resistant viruses, we found that autologous IgG could promote ADCC by NK cells (Figure 6, C–E). Interestingly, at the highest IgG concentration (50 μg/mL), we observed higher ADCC against target cells expressing aNAb-sensitive Envs compared with most of the corresponding participant’s aNAb-resistant Envs (Figure 6, F–H). However, there was no correlation between the neutralization IC50 and ADCC measured as percentage killing at 50 μg/mL IgG (not shown). The mixture of neutralizing and nonneutralizing antibodies in purified IgG samples complicates analysis of the effect of neutralizing antibodies on ADCC, and multiple studies highlight the role of nonneutralizing antibodies in ADCC (89–92). Nevertheless, our results suggest that the ADCC activity of aNAbs may play a role in selection, providing one explanation for the high resistance to aNAbs after 20 years of ART.
 Figure 6
Figure 6Cells infected with outgrowth viruses are susceptible to ADCC with autologous IgG obtained after long-term ART. (A) In vitro ADCC assay. Participant-specific env genes from outgrowth viruses were cloned into NL4-3-ΔNef-eGFP backbone to generate replication-competent reporter viruses. Target CEM.NKR.CCR5 cells (88) were infected with reporter viruses and after 2 days were stained with Cell Trace Violet (CTV). Target cells were cocultured with uninfected donor NK cells and autologous IgG in the presence of antiretroviral drugs (ARVs) and IL-2 for 4 hours. The reduction of infected cells was assessed by flow cytometry. (B) Representative flow cytometry plots showing the percentage of live GFP+ CTV+ CEM.NKR.CCR5 target cells after coculture with uninfected donor NK cells in the presence of the lowest (0.4 μg/mL) and highest concentration (50 μg/mL) of autologous IgG. Plots from control wells with uninfected donor IgG, without IgG, and without NK cells are shown. (C–E) For 3 long-term ART participants, measurements show the percentage of live GFP+ CTV+ CEM.NKR.CCR5 target cells after coculture with uninfected donor NK cells in the presence of autologous IgG. Target cells were infected with GFP-reporter outgrowth viruses (1 color each) and stained with CTV. aNAb-sensitive viruses are indicated in pink with the corresponding aNAb IC50 values, and aNAb-resistant viruses (IC50 values > 100 μg/mL) are represented in other colors. Data are reported as a mean percentage of 2 replicates normalized to uninfected donor IgG. (F–H) Data from the highest autologous IgG concentration (50 μg/mL) in C–E are replotted with the mean and 2 replicates shown. Colors correspond to the same viruses as in C–E.
Large aNAb-resistant outgrowth clones remain stable in size. Given the potential role of aNAbs in selecting against cells carrying sensitive variants, we hypothesized that large clones of infected cells carrying aNAb-resistant viruses might persist in PWH on very long-term ART. To definitively identify infected cell clones, we used matched integration site and near full-length proviral sequencing (MIP-seq) (93). Integration sites were matched to the env sequences of 2 large clones, each from a different PWH on long-term ART in the aNAb-resistant group (Figure 7A). For those 2 viruses, we designed a duplex digital droplet PCR (ddPCR) assay using primers and probes spanning the virus-host junction (integration site) as well as the HIV-1 LTR (R-U5), to determine the frequency of the clones among resting CD4+ T cells (94). One clone from participant SCOPE2046 gave rise to 11 QVOA isolates (Supplemental Figure 2) and was resistant to aNAbs (IC50 of approximately100 μg/mL) from both timepoints tested, after 4.5 and 25.3 years on ART (Figure 5F). The integration site was chr4: 451609(+), within the ZNF721 gene. ZNF genes have been associated with regions of heterochromatin, leading possibly to deeper proviral latency (95, 96). Large clones integrated into ZNF721 have been previously detected in PWH on ART and elite controllers, in some cases with viral outgrowth assays (69, 93, 97–99). Duplex ddPCR analysis on 5 cell samples spanning 11 years of ART showed that the frequency of this clone was stable (average 6.4/million resting CD4+ T cells; Figure 7, B and C). This was 1.1%–2.5% of the average frequency of all proviruses assayed (347 of 1,000,000 resting CD4+ T cells, determined as half of the LTR frequency). For perspective, approximately 90% of proviruses are defective in PWH on ART (100). The average frequency of intact proviruses in this participant was 9.5 copies/million resting CD4+ T cells, as determined by the IPDA (101), using the same cell samples. Thus, on average, this stable resistant outgrowth clone makes up 67% of all intact proviruses, consistent with the highly aNAb-resistant reservoir in this donor (Figure 1B). The second integration site match was for the major outgrowth clone (23 isolates) from SCOPE2256 (Supplemental Figure 2). This clone showed aNAb resistance (IC50 > 100 μg/mL) over 13 years of ART (Figure 5G). The integration site was chr19: 58193728(–), within the ZNF274 gene. Chromosome 19 is enriched with repressive chromatin marks covering ZNF genes (95, 102, 103). Proviral clones integrated in chromosome 19 (19, 69), including ZNF274 (104), have been identified in elite controllers and PWH on ART. In 3 samples spanning 6 years of ART, the frequency of this provirus remained stable (average 61.2/million resting CD4+ T cells; Figure 7, B and C). The average frequency of total proviruses was 1,835 copies/million resting CD4+ T cells, and the average frequency of intact proviruses was 277.2/million resting CD4+ T cells. Thus, this resistant outgrowth clone is a small fraction of all persistent proviruses (2.1%–4.2%) but is a substantial proportion of the intact reservoir, on average nearly 25%. These data support long-term persistence of large aNAb-resistant outgrowth clones that are inducible in the QVOA despite integration into ZNF genes and are potentially capable of causing rebound due to aNAb resistance.
 Figure 7
Figure 7Persistence and prevalence of aNAb-resistant outgrowth clones integrated in ZNF regions. (A) Duplex digital droplet PCR (ddPCR) assays were designed to capture the frequency of proviruses with a specific integration site and env sequence matching an aNAb-resistant outgrowth clone. ddPCR plots display representative data from one well. (B) Longitudinal quantification of the SCOPE2046 ZNF721 and SCOPE2256 ZNF274 proviruses over time on ART. ZNF frequencies (pink or purple) and LTR/2 frequencies (blue) were determined by custom ddPCR assay. Intact proviral frequencies (green) were determined by IPDA (101). Data from 2–16 replicates are shown as proviral copies per 1 × 106 resting CD4+ T cells (mean, SEM) after correction for DNA shearing. ZNF percentages of LTR/2 and intact proviruses are shown above the graph at the corresponding time on ART. Dashes (--) represent ZNF frequencies larger than the intact proviral frequency. (C) Pie charts depicting proportion of ZNF721 (pink) or ZNF274 (purple) out of respective detected LTR/2 copies for each time point.



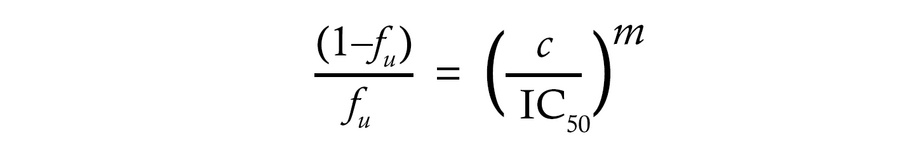
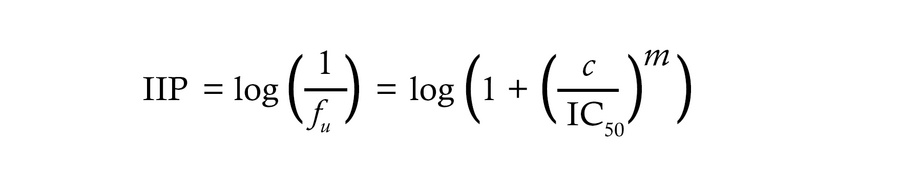







Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)