Advertisement
Research ArticleDermatologyImmunologyNeuroscience
Open Access | ![]() 10.1172/JCI192328
10.1172/JCI192328
Skin-resident Langerhans cells drive neuropathic pain via chemokine-dependent neuron-immune communication
Paola Pacifico,1 Dale George,2 Nirupa D. Jayaraj,1 Dongjun Ren,3 James S. Coy-Dibley,1 Abdelhak A. Belmadani,3 Sofia Veronesi,1 Mirna Andelic,4 Daniele Cartelli,4 Grazia Devigili,4 Raffaella Lombardi,4 Giuseppe Lauria Pinter,4,5 Amy S. Paller,6 Richard J. Miller,3 and Daniela M. Menichella1,3
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Pacifico, P. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by George, D. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Jayaraj, N. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Ren, D. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Coy-Dibley, J. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Belmadani, A. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Veronesi, S. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Andelic, M. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Cartelli, D. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Devigili, G. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Lombardi, R. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Pinter, G. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Paller, A. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Miller, R. in: PubMed | Google Scholar
1Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
2SonoThera Inc., South San Francisco, California, USA.
3Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
4Neuroalgology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy.
6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Address correspondence to: Daniela Maria Menichella, Department of Neurology and Pharmacology, Feinberg School of Medicine, Northwestern University, Lurie 8-123, 303 E. Superior Street, Chicago, Illinois 60611, USA. Phone: 312.503.3223; Email: d-menichella@northwestern.edu.
Find articles by Menichella, D. in: PubMed | Google Scholar
Published April 30, 2026 - More info
J Clin Invest. 2026;136(12):e192328. https://doi.org/10.1172/JCI192328.
© 2026 Pacifico et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: February 12, 2025; Accepted: April 10, 2026
-
Results
Epidermal LC expansion correlates with mechanical allodynia in a mouse model of painful diabetic neuropathy. To explore a possible role of LCs in PDN, we analyzed LC density in the well-established, and clinically relevant, HFD model of PDN (37–39). Consistent with the literature (40), 10 weeks of HFD induced obesity, glucose intolerance, pain behaviors such as mechanical allodynia, and remodeling of cutaneous innervation (37–41). In this model, we observed an increase in LC density in the whole-mount epidermis of wild-type male mice fed an HFD for 10 weeks, compared to those on a regular diet (RD) (Figure 1, A and B).
 Figure 1
Figure 1Increased LC density in HFD male mice. (A and B) Quantification of LC density (number of LCs/mm2) in RD and HFD male mice at 10 weeks (10w) (A) and representative images (B). n = 4 animals for RD and n = 5 animals for HFD. n = 3 sections per animal were acquired and CD207+ cells per 0.04 mm2 per section area were counted. Unpaired 2-tailed t test with Welch’s correction *P = 0.0176. (C) MACSQuant Tyto cell sorting of CD207+/CD45+ gated cells from single cells paw epidermis suspension. PE-CD207 and APC-CD45 antibodies were used to colabel LCs. (D) Graph shows the average percentage from 3 replicates of CD207+/CD45+-sorted cells in RD and HFD. Unpaired 2-tailed t test **P = 0.0010. n = 4 animals per diet condition per each replicate. (E) Time-course experiment of LC density at 2, 4, 6, 8, and 10w show the progressive increase of LC in HFD. Phenotypic characterization of HFD mice at 2, 4, 6, 8, and 10w obtained from published data (38). Two-way ANOVA for multiple comparisons. Between RD and HFD: 8w *P = 0.0216. 10w **P = 0.0014. Within HFD: 2w versus 10w P = 0.0111; 4w versus 10w P = 0.0116; 6w versus 10w P = 0.0486; 8w versus 10w P = 0.0469. 2w: n = 3 animals for both RD and HFD. 4w: n = 3 animals for both RD and HFD. 6w: n = 3 animals for RD and n = 4 for HFD. 8w: n = 4 animals for RD and n = 3 for HFD. 10w: n = 4 animals for RD and n = 3 for HFD. (F) Representative images of LCs CD207+ in epidermal sheets of RD and HFD mice at different time points. (G) Correlation matrix of HFD male features at different time points (2, 4, 6, 8, 10w) including Glucose Tolerance Test (GTT), mechanical allodynia (vonFrey) and LC density. Red squares indicate negative correlations. 10w vonFrey-LCs: Pearson r = –0.9199, P = 0.003. (H) Negative correlation between LC density and mechanical threshold measured with vonFrey test in 10w HFD mice. Pearson r coefficient –0.9199; r2 = 0.8462; P = 0.0033.
To further validate these results, we utilized flow cytometry, a complementary approach commonly used to isolate and quantify immune cells, including LCs (36, 42–45). Consistent with the histological results, flow cytometry revealed a higher proportion of CD45/CD207–double-positive cells in HFD male mice (Figure 1, C and D). Due to the delicate nature of LCs and their low abundance, we used the gentle MACSQuant Tyto sorter (Miltenyi Biotec) to isolate epidermal cells, pooling 3 mice per diet condition for each replicate. The percentage of CD45+/CD207+ cells in HFD was approximately double that of the RD control (Figure 1D).
To determine whether the observed increase in LCs was due to a localized or systemic change resulting from their activation and proliferation, we labeled LCs in whole-mount ears from both RD and HFD mice. Our analysis showed no significant differences in LC density within the ear epidermis (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI192328DS1). This suggests that, as in patients with PDN where the distal lower extremities are primarily affected, the HFD induces a localized increase in LCs in the paw epidermis.
To evaluate the temporal dynamics of LC changes in response to HFD and their contribution to the development of mechanical allodynia, we measured the LC density at different time points after starting either RD or HFD (2 weeks, 4 weeks, 6 weeks, 8 weeks, and 10 weeks). We observed that the LC density gradually increased over time, becoming significantly higher by week 8 (Figure 1, E and F). We have already shown that, at this time, mice on HFD display mechanical allodynia and small fiber degeneration (38). Correlation analyses revealed that, after 10 weeks on HFD, a higher LC density correlated with a lower mechanical threshold, suggesting that an increased number of LCs corresponded to heightened mechanical pain (Figure 1, G and H). We observed a correlation between the number of LCs and the mechanical threshold in male mice fed a RD; although this was within the normal range of mechanical thresholds (Supplemental Figure 1, B and C).
Because sex differences are fundamental biological variables that influence immune cells, particularly microglia (46–48) and, consequently, immune responses (49) and neuropathic pain (18, 50), we decided to investigate the role of LCs in female mice. We first characterized the phenotype in female mice after 10 weeks on the high-fat diet. Like male mice, female mice on HFD developed obesity, glucose intolerance, and mechanical allodynia (Supplemental Figure 1, D–F). However, histology analysis of paw epidermis uncovered no differences in LC density between RD and HFD female mice (Supplemental Figure 1G). Moreover, RD female mice displayed a LC density higher than that of RD male mice. This suggests that female mice have a higher LC density in the epidermis than male mice (Supplemental Figure 1H).
LC morphologic remodeling tracks disease progression in patients with painful diabetic neuropathy. To enhance the clinical relevance of our findings, we analyzed skin biopsies obtained from clinically well characterized male and female patients with PDN and people who were healthy controls. This included samples from 15 patients with PDN and 9 people who were healthy controls (Table 1). In these human skin biopsies, fixed samples, it was not possible to isolate the epidermis, so we performed 3D reconstructions of LCs in PDN skin samples. Our analysis revealed that both the volume of LCs and the complexity of their arborization processes positively correlated with the duration of diabetes in patients with PDN (Figure 2, A–D). Specifically, our findings indicated that, as diabetes progressed, LCs became larger and their branching structures more complex (Figure 2, C and D). As with other immune cells, which exhibit remarkable plasticity, LC morphology is closely related to their function (51), suggesting that structural and functional changes may reflect more severe phenotypes in patients. Consistent with previous studies (3, 4, 30, 33, 52–54), we found that patients with PDN exhibited a significant reduction in intraepidermal nerve fiber (IENF) density compared with controls, while LC density remained unchanged (Supplemental Figure 2). However, the ratio between IENFD and LC density was reduced in both patients with PDN and in the HFD mouse model of PDN (Figure 2, E and F).
 Figure 2
Figure 2LC morphological analysis in patients with PDN. (A) representative images of LCs of patients suffering for diabetes for 5 (left panel) and 16 years (right panel). Scale bar: 15 μm. (B) 3D reconstruction of LCs. (C) Scatter correlation plots between LCs volume and duration of diabetes. Spearmann correlation r = 0.72, P = 0.003. (D) Scatter correlation plots of number of branch points and duration of diabetes. Spearmann correlation r = 0.58, P = 0.028. (E) ratio between the denervation (IENFD), calculated as deviation of the participants IENFD from the normative value corrected for age and sex, and LC density in healthy participants or patients with PDN. Mann-Whitney U test. *P < 0.05. Middle lines of the plots represent the mean value, while whiskers are SEM. Right panels show representative confocal micrographs of epidermal nerve fibers PGP 9.5+ (green) and LCs CD207+ (red) in skin sections of healthy participants and patients with PDN. Scale bar: 50 mm. (F) Ratio between intraepidermal nerve fibers density and LC density is significantly decreased in HFD male mice. Unpaired 2-tailed t test with Welch’s correction **P = 0.0066. Right panels. Representative confocal images (low-magnification) of nociceptive fibers Nav1.8-positive (green) and LCs CD207+ (red) in skin sections of RD and HFD male mice.
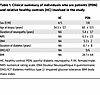 Table 1
Table 1Clinical summary of individuals who are patients (PDN) and relative healthy controls (HC) involved in the study
Targeted depletion of epidermal LCs blocks mechanical allodynia and spontaneous pain in PDN mice. To functionally test in vivo the requirement of LCs for PDN development, we ablated LCs using a transgenic mouse line expressing the diphtheria toxin receptor (DTR) under the control of the langerin/Cd207 promoter (36, 55). We administered diphtheria toxin (DT) observing that weekly injections, from week 4 to week 8, effectively depleted LCs in DT-treated mice (Figure 3A). Indeed, histological analysis of paw epidermal sheets confirmed a 70% reduction in LC density 2 days after the final DT injection compared with control mice injected with vehicle (0.9% NaCl) (Figure 3, D and E).
 Figure 3
Figure 3LCs mediate mechanical allodynia and spontaneous pain in HFD. (A) Timeline of diphtheria-toxin ablation strategy in hLC-DTR HFD male mice. (B) Response to evoked mechanical stimuli threshold. von Frey test shows the recovery of mechanical allodynia in HFD DT-ablated mice. One-way ANOVA for multiple comparison: RD (vehicle) versus HFD (vehicle) ****P < 0.0001; RD (vehicle) versus RD (DT) P = 0.9995 (ns); RD (vehicle) versus HFD (DT) P = 0.0514 (ns); HFD (vehicle) versus HFD (DT) *P = 0.0213; HFD (vehicle) versus RD (DT) ***P = 0.0001; HFD (DT) versus RD (DT) P = 0.0658 (ns). RD (vehicle) n = 6; RD (DT) n = 6; HFD (vehicle): n = 6 HFD (DT): n = 8. (C) Spontaneous pain behavior measured as time to fall (seconds) is fully recovered by DT-ablation in HFD. One-way ANOVA for multiple comparison: RD (vehicle) versus HFD (vehicle) ***P = 0.0001; RD (vehicle) versus RD (DT) P = 0.9824 (ns); RD (vehicle) versus HFD (DT) P = 0.9907 (ns); HFD (vehicle) versus HFD (DT) ***P = 0.0003; HFD (vehicle) versus RD (DT) ****P < 0.0001; HFD (DT) versus RD (DT) P = 0.9088 (ns). RD (vehicle) n = 7; RD (DT) n = 7; HFD (vehicle): n = 8 HFD (DT): n = 7. (D) Representative confocal images of paw epidermal sheets of HFD male mice injected with vehicle (0.9% NaCl) or DT 4n g/gr body weight. (E) LC density measured as number of CD207+ cells/area. Unpaired 2-tailed t test with Welch’s correction ***P = 0.0003. HFD (vehicle): n = 4. HFD (DT): n = 3. (F) Representative images of H&E (low-magnification) labeled skin sections of HFD male mice vehicle- and DT- injected. (G) quantification of epidermal thickness in HFD mice vehicle- and DT- injected. Unpaired 2-tailed t test with Welch’s correction P = 0.1681 (ns).
Behavioral testing revealed that repeated DT injections prevented the development of mechanical allodynia in the HFD male mice (Figure 3B). In contrast, HFD mice that received vehicle treatment maintained robust allodynia (Figure 3B), indicating that LCs are necessary for the development of mechanical allodynia in the HFD model of PDN. On the contrary, DT-induced LC ablation did not affect the response to mechanical stimulation in RD mice (Figure 3B). Additionally, assessment of spontaneous pain through a cage-lid hanging test (56) showed that HFD male mice developed spontaneous pain, which was reversed by LC ablation (Figure 3C). Histological analysis confirmed that the DT-mediated strategy effectively removed LCs (Figure 3, D and E) without altering the histological organization of the epidermis (Figure 3, F and G). Quantification of IENF density of PGP9.5-positive fibers revealed that LC depletion could potentially influence cutaneous fiber innervation in HFD male mice, although no statistically significant differences were detected (Supplemental Figure 3A). To assess whether LC loss affected a defined subset of DRG sensory neurons previously linked to these cells (36), we generated a double-transgenic mouse line by crossing Mrgprd-eGFP mouse line (57) with hLC-DTR mice (MrgprdeGFP-hLCDTR). As observed in hLC-DTR mice, DT-mediated LC depletion in MrgprdeGFP-hLCDTR mice prevented the development of HFD-induced mechanical allodynia (Supplemental Figure 3, B and C), without affecting dermal cell populations (Supplemental Figure 3D). Quantification of IENF density of Mrgprd-eGFP-positive fibers revealed no significant differences between DT- and vehicle-treated mice (Supplemental Figure 3E), highlighting the difficulty of capturing dynamic processes as fiber degeneration and regeneration at this PDN stage.
We also evaluated the impact of LC depletion in female HFD mice. We found that, as opposed to males, in female mice, the loss of LCs induced by DT did not prevent mechanical allodynia (Supplemental Figure 3, F and G), suggesting that sex-dependent mechanisms influence the role of LCs in PDN. Together, these results demonstrated that epidermal LCs are essential for maintaining mechanical allodynia and spontaneous pain in the male HFD model of PDN.
Single-cell transcriptional profiling reveals sexually dimorphic signatures in mouse epidermis. To investigate the molecular mechanisms underlying LC-dependent neuropathic pain in the HFD mouse model of PDN, we performed an unbiased single-cell RNA-seq (scRNA-seq) of the paw epidermis in male mice. Mice were fed either an RD (RD n = 3) or an HFD (HFD n = 3) for a duration of 10 weeks (Figure 4A). After separating the epidermis from the dermis, we obtained a high-viability single cell–suspension from each sample, collecting 40,500 cells from the RD epidermis and 28,950 cells from the HFD epidermis. Single cells were sequenced using the 10x Genomics Chromium platform. To remove doublets and low-quality cells, we filtered out cells with fewer than 200 or more than 6,000 features and those containing more than 5% mitochondrial reads (Supplemental Figure 4, A–G). We identified 10 distinct clusters based on the shared nearest neighbor (SNN) clustering algorithm in Seurat. We then visualized the different clusters using the 2-dimensional Uniform Manifold Approximation and Projection (UMAP) method (Figure 4B). Based on known marker expression and the top 5 differentially expressed genes (DEGs) for each cluster (Figure 4C), we identified 8 keratinocyte subpopulations representing different stages of differentiation and 2 groups of nonkeratinocytes. As keratinocytes are the most abundant cell type in the epidermis, we used multiple markers to define their differentiation stages (Figure 5 and Supplemental Figure 5A). Feature plots illustrated the distribution of keratinocyte markers, highlighting terminally differentiated keratinocytes, usually located in the upper layer of the epidermis corresponding to the stratum corneum, differentiated keratinocytes including spinous, suprabasal, and squamous keratinocytes, undifferentiated basal epithelium keratinocytes mainly expressing Keratin14 (Krt14), migratory keratinocytes and keratinocytes with higher expression of Mki67,a distinctive marker of proliferation (Figure 5 and Supplemental Figure 5, A and B). Specifically, we identified: (a) terminally differentiated keratinocytes (tdKCs) expressing Flg, Krt78, Krt80, Lor and Ivl; (b) spinous keratinocytes (sKCs) expressing Cstdc5 and Dsg1a; (c) suprabasl keratinocytes type I (sbKCs I) and (d) type II (sbKCs II) specified by Krt10 and Krt1, and Krt16, Krt17 and Il34, respectively; (e) squamous keratinocytes (sqKCs) expressing Krt16, Serpinb12 and Tmem266. To define undifferentiated KCs, we used Krt14, Itga6 and Itgb1 for (f) basal layer keratinocytes (blKCs); Krt79, Dcn, Lrig1 and Sema3e for (g) migratory Keratinocytes (mKCs) and, lastly, Mki67 and Cenpa for (h) proliferating keratinocytes (pKCs) (Figure 5). Furthermore, we used the high and selective expression of Cd207, Cd74, Cd28, and Cd3e to identify the remaining no-keratinocytes as LCs (i) and immune cells (j), respectively (Figure 5).
 Figure 4
Figure 4scRNA-seq of paw epidermis of RD and HFD male and female mice. (A) Schematic workflow of scRNA-seq of paw epidermis of RD and HFD male and female mice. (B) UMAP plot visualization of all 10 clusters identified in scRNA-seq data of RD and HFD male paw epidermis. (C) Heatmap of top 5 differentially expressed genes in each cluster. (D) UMAP dimensionality reduction of RD and HFD scRNA-seq paw epidermis of female mice shows 10 distinct clusters identified by the expression of differentially expressed genes (DEGs). (E) Heatmap of top 5 DEGs expressed in each cluster. (F and G) UMAP dimensionality reduction of integration analysis of RD and HFD both male and female scRNA-seq datasets using anchor-based CCA integration. (H) Enrichment pathways analysis using EnrichR comparing HFD LCs in male and female datasets.
 Figure 5
Figure 5Profiling of epidermal cell clusters. Feature plot of the most representative gene markers used to identify each epidermal cluster. Terminally differentiated Keratinocytes (tdKCs) identified by Flg, Krt78, Krt80, Lor, and Ivl. Spinous Keratinocytes (sKCs) identified by Cstdc5 and Dsg1a. Suprabasl keratinocytes type I (sbKCs I) identified by Krt10 and Krt1. Suprabasal Keratinocytes type II (sbKCs II) identified by Krt16, Krt17, and Il34. Squamous Keratinocytes (sqKCs) identified by Krt16, Serpinb12, and Tmem266. Basal Layer Keratinocytes (blKCs) identified by Krt14, Itga6, and Itgb1. Migratory Keratinocytes (mKCs) identified by Krt79, Dcn, Lrig1 and Sema3e. Proliferating Keratinocytes (pKCs) identified by Mki67 and Cenpe. Nonkeratinocyte cells LCs identified by Cd207 and Cd74; Immune Cells identified by Cd3e and Cd28.
Next, we extended the single-cell transcriptomic analysis to the epidermis of female mice in the HFD model of PDN. We performed scRNA-seq of the paw epidermis of female RD (RD n = 3) and HFD (HFD n = 3) mice (Figure 4D). A high-viability single cell–suspension containing 33,175 cells from RD epidermis and 31,047 cells from HFD epidermis, was analyzed for quality control and filtration steps to improve data reliability and exclude low-quality or dying cells (Supplemental Figure 4, H and I). Like male scRNA-seq data, 10 clusters were identified in female mice, primarily keratinocytes, immune cells, and LCs (Figure 4, D and E). To assess sex-dependent molecular differences, we conducted a comprehensive comparison of male and female scRNA-seq datasets under both RD and HFD conditions. We integrated both scRNA-seq datasets while correcting for batch effects across sexes and diet conditions, thereby improving comparability between samples (Figure 4F). The integrated UMAP analysis revealed extensive overlap between male and female datasets, indicating a strong correspondence and comparable representation of the distinct clusters across sexes (Figure 4G and Supplemental Figure 4J). Focusing on the LC cluster, we performed a web-based gene set analysis (58, 59) to identify diet-enriched pathways in both sexes. We found that the LC cluster showed upregulation in pathways linked to protein translation and synaptic signaling only in male HFD mice (Figure 4H).
Finally, we validated selected cluster markers in paw skin sections of RD and HFD mice. KRT10 was used as a marker of epithelial differentiation, while KRT14 labeled basal cells in contact with the basement membrane (Supplemental Figure 4K). Epidermal LCs were labeled using CD207 in paw skin sections from RD and HFD male mice (Supplemental Figure 4L).
Transcriptional profiling reveals inflammatory and axonal guidance programs in LCs from PDN male mice. To define the transcriptional programs altered across epidermal cell types in the HFD model of PDN, we performed a comparative clustering analysis of scRNA-seq datasets from RD- and HFD-fed mice. This approach revealed shifts in molecular features across several epidermal populations in male mice (Figure 6A). Although the integrated male-female analysis showed comparable overall epidermal cellular composition, LCs emerged as the most sex-divergent cell type under HFD conditions (Figure 4, F and G). Male LCs, but not female LCs, exhibited robust transcriptional remodeling in response to diet. We therefore focused subsequent analyses on defining the pathways specifically altered in LCs from PDN male mice fed a HFD compared with RD-fed controls.
 Figure 6
Figure 6GSEA of LC cluster. (A) UMAP plots of the scRNA-seq of the 2 diet conditions, RD and HFD. (B) Plot shows GSEA of Langerhans cells cluster identifies enriched sets of genes with positive and negative NES value (|NES| > 1.88). (C–E) examples of major enriched pathways with positive NES showing the enrichment in MHC-II–related genes (C) and negative NES (D–E. Statistics were derived using the fgsea package in R. Adjusted P value results from Benjamini-Hochberg correction. (F and G) Stacked violin plot showing the expression of genes implicated in axonal guidance, such as Plxna1 and Plxnb2, and inflammatory response. Combined (F) and split (G) by diet conditions gene expression (RD, blue; HFD, red).
Gene set enrichment analysis (GSEA) (60) using the Mouse MSigDB (61) on Seurat-derived DEGs (see methods) identified discrete functional programs enriched in male HFD LCs. Pathways with positive normalized enrichment scores (NES) were dominated by MHC class II–mediated antigen-presentation signatures, whereas pathways with negative NES values mapped to synaptic protein networks and inflammatory signaling modules (Figure 6, B–E, and Supplemental Figure 6, A and B).
Integration of these transcriptional signatures with prior literature (62) further highlighted coordinated upregulation of immune-response genes and axonal-guidance pathways in male HFD LCs. Notably, the axon-guidance receptors Plxnb2 and Plxna1 were significantly increased, along with chemokine-related and other immune signaling genes (Figure 6, F and G). These findings indicate that HFD exposure in PDN males elicits a combined antigen-presenting, inflammatory, and neuroimmune remodeling program in LCs.
In contrast, the LC cluster from female HFD mice exhibited a nonoverlapping DEG profile characterized by increased expression of the sodium channel Scn3a and the purinergic receptor P2rx2 (Supplemental Figure 6C). Comparative analysis of male and female HFD datasets confirmed that LC transcriptional responses to diet are highly sex specific and involve distinct molecular pathways (Supplemental Figure 6E).
Given the known cellular and functional diversity of LCs (45, 63–65), we conducted a subcluster analysis of the major cluster of these cells. This analysis revealed 4 transcriptionally distinct subtypes (Figure 7A), each characterized by unique markers (Figure 7B and Supplemental Figure 7, A and B). Each of these LC subtypes may carry out different functions in PDN. We identified (a) Epcam+/Cd48+ LCs, (b) Ly6g6c+ LCs, (c) Cenpe+ LCs, and (d) Ccr7+ LCs. All 4 clusters expressed the common LC marker Cd207 (Figure 7, A–C). Specifically, Epcam+/Cd48+ LCs were identified by the expression of Cd48, which was elevated in HFD (Supplemental Figure 7, A and B). Cd48, a member of the signalling lymphocyte activation molecule family, is usually associated with heightened activation of immune cells (66), including LCs (44). Ly6g6c+ LCs indicated a monocyte-derived origin (Figure 7E), and, similar to resident macrophages, Ly6g6c+ LCs might be recruited from the blood, contributing to the higher number of cells observed in the HFD epidermis (63, 67). Cenpe+ LCs (Supplemental Figure 7, A and B) suggested that a small number of LCs might undergo local cell division and proliferation (68). Ccr7+ LCs represented a migratory phenotype (42, 69) (Figure 7D). We also detected the transcripts of Tnfa, Tgfb1, Ccl22 (Supplemental Figure 7, A and B), and Lpar3, markers of nonpeptidergic nociceptors type 1 (NP1) (70, 71), which were upregulated in HFD (Figure 7F), as well as Ramp1, coreceptor of calcitonin gene-related peptide receptor (CGRP) (72) (Figure 7G). HFD LCs subtypes also expressed high levels of axonal guidance molecules such as Plxnb2 and Plxna1 (Figure 7, I and J). Moreover, we found that inflammatory mediators, specifically IL-18 (Figure 7H) and the chemokine Ccl2, were upregulated in HFD LCs (Figure 7, K and L). Notably, these inflammatory mediators, especially CCL2, have been implicated in mediating neuropathic pain (73, 74).
 Figure 7
Figure 7LC subclustering analysis and gene expression profiles. (A) UMAP plots of the LC subclustering showing 4 distinct groups. (B) Stacked violin plot of marker genes adopted to identify LC subclusters. (C–H) ViolinPlot showing the expression of Cd207 (C), Ccr7 (D), Ly6g6c E), Lpar3 (F), Ramp1 (G) and IL18 (H) in LCs subclusters. (I–L) ViolinPlots show the expression and distribution of plexins molecules Plxnb2 (I) and Plxna1 (J) and chemokine molecules (K) Mcp1/Ccl2 and (L) Mip-1b/Ccl4 in LC subclusters.
In female LCs, we identified 4 subtypes: (a) Epcam+/Cd48+ LCs, (b) Ly6g6c + LCs, (c) Ccr7+ LCs, and (d) Cenpe+ LCs, which are similar to the subtypes found in males (Supplemental Figure 7, C and D). However, axonal guidance molecules such as Plxnb2 and Plxna1, as well as the chemokine CCL2, were not detected (Supplemental Figure 7E). This indicates that female LCs may interact with sensory fibers through different mechanisms.
Ligand-receptor analysis reveals neuro-immune crosstalk via Semaphorin-plexin and chemokine pathways. To investigate how LCs communicate with surrounding cells and nerve afferents in the epidermis, we applied 2 complementary computational platform analyses to our scRNA-seq datasets. We used CellChat (75) and the interactome analysis platform (76) to infer ligand-receptor–mediated signaling (Figure 8A). Focusing on the male dataset, CellChat indicated that both the number of inferred cell-cell communications and the strength of these interactions were increased in HFD mice compared with RD controls (Supplemental Figure 8, A and B). Pathway analysis showed dramatic differences between RD and HFD conditions, including among keratinocyte clusters with changes in the pleiotrophin (Ptn) or Epidermal Growth Factor (Egf) pathways, which are known to be upregulated in wound repair and healing (77), or in cell adhesion molecules (Nectin) (Figure 8B). Moreover, among pathways involved in neuronal communication, semaphorin signaling, especially Sema6 and Sema4 families, was differentially regulated in HFD LCs (Figure 8, C and D). Integrating epidermal scRNA-seq data with DRG (57) datasets from RD and HFD revealed upregulation of Plxnb2 and Plxna1 receptors in HFD LCs, while their ligands, Sema4d and Sema6d, respectively, were expressed by HFD nonpeptidergic nociceptor type 1 (NP1) DRG neurons (Supplemental Figure 8, C–F). Further analysis of LC subclusters and NP1 DRG cells expressing Mrgprd indicated that communication between LCs and NP1 also relies on synaptic contact molecules, such as neurexins (Nrxn) (Figure 8, E–H, and Supplemental Figure 8, G–J), suggesting that LCs may establish synaptic-like contacts with sensory afferents in the epidermis. Using the interactome analysis platform (76) with LCs as the receptor component, we confirmed that these cells communicate with NP1 DRG through Sema4- and Sema6-signaling pathways (Figure 8I). In-situ RNA scope in DRG sections also confirmed the expression of Sema4d and Sema6d in NP1 Mrgprd+ DRG neurons (Supplemental Figure 8, K and L), supporting putative LC-NP1 communication through these pathways. Interestingly, when the ligand-receptor expression was inverted, LCs were predicted to interact with NP1 neurons via the chemokine CCL2 pathway (Figure 8J).
 Figure 8
Figure 8Cell-cell communication between LCs and DRG sensory neurons. (A) Schematic cartoon of cell-cell ligand-receptor network analysis performed using CellChat and Interactome analysis platforms (B) Comparison of cell-cell communication between RD and HFD using Heatmap plot. Highlighted in bold Sema4 and Sema6 signaling pathways (C and D) Circle plots show Sema4 and Sema6 signaling pathways in RD and HFD. (E–H) Chord grams of integrated analysis of HFD LC subclusters and HFD NP1 DRG. (I and J) interactome analysis platform to explore the communication between HFD LC cluster and HFD NP1 DRG.
Langerhans cells modulate nociceptive responses via CCL2-CCR2 chemokine signaling. As key players in the innate immune response (26), LCs respond to activation by releasing proinflammatory cytokines and nitric oxide, factors that can sensitize epidermal DRG axonal afferents (18). To define how LC secretory programs are altered in PDN, we performed multiplexed proteomic profiling on sorted CD45+/CD207+ LCs from RD and HFD male mice (Figure 9A). Consistent with their immunologic function, LCs secreted TNFA, GMCSF, IL-10, and MIF (Supplemental Figure 9, A and B). Strikingly, monocyte chemoattractant protein-1 (MCP-1/CCL2) remained elevated in HFD LCs following TNFA stimulation for 24 hours (Figure 9B), confirming at the protein level that chemokine CCL2 is upregulated in HFD-associated LC dysfunction. In contrast, macrophage inflammatory protein-1β (MIP-1β/CCL4) was selectively secreted by RD LCs under the same inflammatory conditions (Figure 9C). We also identified dysregulation of platelet-derived growth factor-BB (PDGF-BB), a potent mitogenic signal, in HFD LCs (Figure 9D), highlighting broader disruption of LC secretome pathways in PDN.
 Figure 9
Figure 9High-multiplexing cytokine profile of LCs in RD and HFD. (A) Representative workflow of cytokine profile assay (B–D) Barplots show inflammatory molecules detected in the multiplexed proteomic profiling. Black arrows indicate differences for diet and treatment condition. (E and F) Intraplantar injection of CCR2RA improves mechanical allodynia in HFD male mice. Mixed effects analysis for multiple comparisons HFD +CCR2RA: baseline versus 1h ***P = 0.0001, baseline versus 2h *P = 0.0176 HFD vehicle n = 6, HFD CCR2RA n = 8.
Given the well-defined role of CCL2-CCR2 signaling in neuropathic pain (73, 74), we next evaluated its causal contribution to PDN. Local intraplantar administration of a selective CCR2R antagonist (CCR2RA) transiently but robustly reversed mechanical allodynia in HFD mice for approximately 2 hours (Figure 9, E and F), without affecting LC density (Supplemental Figure 9C). Together, these findings demonstrate that peripheral CCL2-CCR2 signaling is required for the development of mechanical allodynia in the HFD model of PDN and identify LCs as an essential source of this pronociceptive chemokine.













Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)