Comments for:

Abstract
Hyperproliferation of the colonic epithelium, leading to expansion of colonic crypt progenitors, is a recognized risk factor for colorectal cancer. Overexpression of progastrin, a nonamidated and incompletely processed product of the gastrin gene, has been shown to induce colonic hyperproliferation and promote colorectal cancer in mice, but the mechanism of pathogenesis has not been defined. Cholecystokinin-2 receptor (CCK2R) is the primary receptor for cholecystokinin (CCK) and amidated gastrin. Here, we show that Cck2r was expressed in murine colonic crypts and upregulated in the transgenic mice that overexpress human progastrin. Murine deletion of Cck2r abrogated progastrin-dependent increases in colonic proliferation, mucosal thickness, and β-catenin and CD44 expression in the colon tumor. In addition, either deletion or antagonism of Cck2r resulted in the inhibition of progastrin-dependent increases in progenitors expressing doublecortin and CaM kinase–like-1 (DCAMKL1), stem cells expressing leucine rich repeat–containing G protein–coupled receptor 5 (LgR5), and colonic crypt fission. Furthermore, in the azoxymethane mouse model of colorectal carcinogenesis, Cck2r deletion in human progastrin–overexpressing mice resulted in markedly decreased aberrant crypt foci formation and substantially reduced tumor size and multiplicity. Taken together, these observations indicate that progastrin induces proliferative effects, primarily in colonic progenitor cells, through a CCK2R-dependent pathway. Moreover, our data suggest that CCK2R may be a potential target in the treatment or prevention of colorectal cancer.
Authors
Guangchun Jin, Vigneshwaran Ramanathan, Michael Quante, Gwang Ho Baik, Xiangdong Yang, Sophie S.W. Wang, Shuiping Tu, Shanisha A.K. Gordon, David Mark Pritchard, Andrea Varro, Arthur Shulkes, Timothy C. Wang
Annexin-II, and not CCK2R, binds progastrin, and mediates biological effects of progastrin/gastrin peptides.
Submitter: Pomila Singh | posingh@utmb.edu
Authors: Dr. Shubashish Sarkar
University of Texas Medical Branch
Published December 18, 2009
We have read the article by Jin, Wang, and colleagues, titled “Inactivating cholecystokinin-2 receptor inhibits progastrin-dependent colonic crypt fission, proliferation, and colorectal cancer in mice” (J. Clin. Invest. (2009);119(9):2691-2701, and wish to draw attention to puzzling aspects of the study in light of previous findings, and correct inaccurate statements regarding our work. In the 1990s, we and others reported expression of the gastrin gene and progastrin, but not amidated gastrins, in colon cancer cell lines and adenocarcinomas (1–3; reviewed in ref. 4). Proliferative, anti-apoptotic, and co-carcinogenic effects of progastrins on intestinal epithelial cells/colonic crypts cells were confirmed by several laboratories, including ours (4–11). The fact that amidated gastrins and cholecystokinin (CCK) bind the CCK-2 receptor (CCK2R) with high affinity, while glycine-extended gastrins and progastrins do not (4,6,12–14), remains undisputed and was acknowledged by Jin et al. in the current study. A large body of evidence has further confirmed that receptors other than CCK2R mediate biological effects of progastrins (4–6,12–15). Cell lines negative for CCK2R respond to proliferative/anti-apoptotic effects of progastrin (4,6,13,15–18), providing evidence that CCK2R are not required for measuring biological effects of progastrin. Using proteomic methods, we identified annexin-II as the non-conventional high-affinity binding ‘receptor’ for progastrin/gastrin peptides (18). Binding of progastrin with annexin-II was confirmed by co-immunoprecipitation and in vitro solution assays (18). In order to avoid contamination with mesenchymal cells, we used isolated colonic crypts from transgenic mice overexpressing progastrin in intestines (Fabp-PG mice) and reported co-localization of progastrin with annexin-II in vivo (10). Extracellular annexin-II associated with outer membranes of cancer and other cells, has been reported (19,20), and confirmed by us in published abstracts. The latter facts were inaccurately presented by Wang and colleagues. Dr. Wang is a co-author of previous publications from our group and Dr. Mark D. Pritchard’s group, in which elevated amidated gastrins in INSulin-GAStrin gene (INS-GAS) mice were reported to have significantly different effects compared to elevated progastrins in hGAS mice (overexpressing progastrin from liver) (4,7,8,21), strongly suggesting that CCK2R does not mediate progastrin effects. In 2008, Dr. Wang’s group reported CCK2R-independent binding of progastrin with unknown glycosaminoglycan proteins on colonic-crypt/intestinal epithelial cells (22); they now report that CCK2R is required for mediating co-carcinogenic effects of progastrin. Thus, the current conclusion of the authors disagrees with previous publications from other laboratories and their own laboratory.
Other concerns not addressed/misrepresented in the current paper include: known pro-apoptotic effects of gastrin-activated wtCCK2R (21,23); debatable expression of wtCCK2R by colonic tumors (4,24); and misrepresentation of our findings with gastrin-knockout mice (25). Finally the data should be discussed in the context of the model of carcinogenesis. The authors used an aggressive, multiple-initiation carcinogeneisis model, rather than an initiation-progression model, with unconventional results. Our understanding of a role of progastrin/splice-variant vs. wtCCK2R/annexin-II in colon tumorigenesis continues to evolve. Since no laboratory, including that of Dr. Wang, has demonstrated binding/co-localization of progastrin with CCK2R, in vitro or in vivo, an indirect role of CCK2R in vivo, if any, needs to be discussed in relation to possible expression of CCK2R/CCK1R by mesenchymal cells.
References
- Van Solinge WW, Nielsen FC, Friis-Hansen L, Falkmer UG, Rehfeld JF. 1993. Expression but incomplete maturation of progastrin in colorectal carcinomas. Gastroenterology. 104:1099-107.
- Singh P, Xu Z, Dai B, Rajaraman S, Rubin N, Dhruva B. 1994. Incomplete processing of progastrin expressed by human colon cancer cells: role of noncarboxyamidated gastrins. Am J Physiol.;266:G459-G468.
- Siddheshwar RK, Gray JC, Kelly SB. 2001. Plasma levels of progastrin but not amidated gastrin or glycine extended gastrin are elevated in patients with colorectal carcinoma. Gut. 48: 47-52.
- Rengifo-Cam, W and Singh, P. 2004. Role of progastrins and gastrins and their receptors in GI and pancreatic cancers: targets for treatment. Curr. Pharm. Des. 10(19): 2345–5238.
- Baldwin GS, Hollande F, Yang Z, Karelina Y, Paterson A, Strang R, Fourmy D, Neumann G, Shulkes A. 2001. Biologically active recombinant human progastrin(6-80) contains a tightly bound calcium ion. J Biol Chem. 276: 7791-7796.
- Singh P, Lu X, Cobb S, Miller BT, Tarasova N, Varro A, Owlia A. 2003. Progastrin1-80 stimulates growth of intestinal epithelial cells in vitro via high-affinity binding sites. Am J Physiol Gastrointest Liver Physiol. 284:G328-G339.
- Singh P, Velasco M, Given R, Varro A, Wang TC. 2000. Progastrin expression predisposes mice to colon carcinomas and adenomas in response to a chemical carcinogen. Gastroenterology. 119:162-171.
- Singh P, Velasco M, Given R, Wargovich M, Varro A, Wang TC. 2000. Mice overexpressing progastrin are predisposed for developing aberrant colonic crypt foci in response to AOM. Am J Physiol Gastrointest Liver Physiol. 278:G390-G399.
- Cobb S, Wood T, Ceci J, Varro A, Velasco M, Singh P. 2004. Intestinal expression of progastrin significantly increases colon carcinogenesis in transgenic mice in response to AOM. Cancer 100:6 1311-1323, 2004.
- Umar S, Sarkar S, Cowey S, Singh P. 2008. Activation of NF-kB is required for mediating proliferative and antiapoptotic effects of progastrin on proximal colonic crypts of mice, in vivo. Oncogene. 27: 5599-5611.
- Umar S, Sarkar S, Wang Y, Singh P. 2009. Functional cross-talk between b-catenin and NFkB signaling pathways in colonic crypts of mice in response to progastrin. J. Biol. Chem. 284:22274–22284.
- Seva C, Dickinson CJ, Yamada T. 1994. Growth-promoting effects of glycine-extended progastrin. Science. 265: 410-412.
- Singh P, Owlia A, Espeijo R, Dai B.1995 Novel gastrin receptors mediate mitogenic effects of gastrin and processing intermediates of gastrin on Swiss 3T3 fibroblasts. Absence of detectable cholecystokinin (CCK)-A and CCK-B receptors. J Biol Chem. 270: 8429-8438.
- Rengifo-Cam, W., Umar, S., Sarkar, S., and Singh, P. 2007. Antiapoptotic effects of progastrin on pancreatic cancer cells are mediated by sustained activation of nuclear factor-kB. Cancer Res. 67: 7266–7274.
- Bold RJ, Ishizuka J, Townsend CM Jr, Thompson JC. 1994. Gastrin stimulates growth of human colon cancer cells via a receptor other than CCK-A or CCK-B. Biochem Biophys Res Commun. 202:1222-1226.
- Wu H, Rao GN, Dai B, Singh P. 2000. Autocrine gastrins in colon cancer cells Up-regulate cytochrome c oxidase Vb and down-regulate efflux of cytochrome c and activation of caspase-3. J Biol Chem. 275:32491-32498.
- Wu H, Owlia A, Singh P. 2003. Precursor peptide progastrin(1-80) reduces apoptosis of intestinal epithelial cells and upregulates cytochrome c oxidase Vb levels and synthesis of ATP. Am J Physiol Gastrointest Liver Physiol. 285:G1097-G1110.
- Singh, P., Wu, H., Clark, C., and Owlia, A. 2007. Annexin II binds progastrin and gastrin-like peptides, and mediates growth factor effects of autocrine and exogenous gastrins on colon cancer and intestinal epithelial cells. Oncogene. 26: 425–440.
- Ortiz-Zapater E. et al. 2007. Tissue plasminogen activator induces pancreatic cancer cell proliferation by a non-catalytic mechanism that requires extracellular signal-regulated kinase 1/2 activation through epidermal growth factor receptor and annexin A2. Am. J. Pathol. 170:1573–1584.
- Shiozawa Y, Havens AM, Jung Y, Ziegler AM, Pedersen EA, Wang J, Wang J, Lu G, Roodman GD, Loberg RD, Pienta KJ, Taichman RS. 2008. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell Biochem. 105:370–380.
- Przemeck SM, Varro A, Berry D, Steele I, Wang TC, Dockray GJ, Pritchard DM. 2008. Hypergastrinemia increases gastric epithelial susceptibility to apoptosis. Regul Pept. 146:147-156.
- Dubeykovskiy, A, Nguyen, T, Dubeykovskaya, Z, Lei, S, and Wang, TC. 2008. Flow cytometric detection of prograstrin interaction with gastrointestinal cells. Regul. Pept. 151:106–114.
- Sebens Müerköster, S et al. 2008. The apoptosis-inducing effect of gastrin on colorectal cancer cells relates to an increased IEX-1 expression mediating NF-_B inhibition. Oncogene. 27:1122–1134.
- Reubi JC, Schaer JC, Waser B. 1997. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 57:1377-1386.
- Cobb S, Wood T, Tessarollo L, Velasco M, Given R, Varro A Tarasova N, Singh P. 2002. Deletion of functional gastrin gene markedly increases colon carcinogenesis in response to azoxymethane in mice. Gastroenterology. 123:516–530.
The Authors’ Reply
Submitter: Timothy Wang | Tcw21@columbia.edu
Authors: Dr. Guangchun Jin, Dr. D. Mark Pritchard
Columbia University Medical Center
Published December 18, 2009
We thank Dr. Pomila Singh for her interest in our work and her many useful and stimulating comments regarding our recent publication (1). As outlined in her letter, Dr. Singh has had a longstanding interest in progastrin and has made a number of important contributions to the field. She has in her letter highlighted a number of salient points, including the fact that progastrin does not bind to the CCK2R with the same affinity as amidated gastrin (and cholecystokinin), and that previous studies (including our own) have suggested that progastrin may bind to cell surface receptors other than CCK2R. We would agree. In addition, she has pointed to possible effects of progastrin on non-epithelial cells (e.g. mesenchymal cells), and this is an active area of investigation by one of our co-authors (D.M.P.). When we began the studies that led to this recent publication (1), we performed the experiments with the stated purpose of showing that CCK2R was not involved and thus we were as surprised as anyone that CCK2R was essential for the proliferative response to progastrin in vivo. However, the requirement for CCK2R in the proliferative and carcinogenic responses to progastrin and azoxymethane (AOM) was shown both using a highly specific CCK2R antagonist and genetically via CCK2R-knockout mice, the latter studies of which were highly reproducible and incontrovertible. With respect to the AOM-induced colon carcinogenesis model, the regimen employed was quite similar to that reported although the dose of AOM used (10 mg/kg/week) was less than that used in earlier studies by Singh et al. (12 mg/kg/week), although the duration of treatment was longer (6 weeks versus 3 weeks) in order to insure a sufficient tumor burden that could be inhibited (2).
However, much of the focus of concern appears to be whether progastrin binds directly to CCK2R. In our recent publication (1), we did not make this specific claim and indeed acknowledged that several earlier studies suggested that this was not the case. However, since this point has been raised, we do need to question whether the few earlier studies that reached this conclusion effectively excluded a direct interaction. We found in our hands that many cell lines that have been reported to show proliferative effects to progastrin, such as IEC-6 and IEC-18 intestinal epithelial cells (3), actually do express CCK2R (Figure 1). In a recent study, rat-specific CCK2R primers (Forward primer: CCTTCTCAACAGCAGTAGTGCCGG; Reverse primer: GGGAGCGTGTTTGCCATACTCGTG) were used that were designed to specifically amplify a 465bp cDNA fragment after reverse transcriptase (that can be distinguished from the 7850bp genomic fragment). Using these primers in RT-PCR assays, the specific 465 bp band was observed in both IEC-6 and IEC-18 cells (ATCC), and sequencing of these bands confirmed that they represent authentic rat CCK2R cDNA sequences.
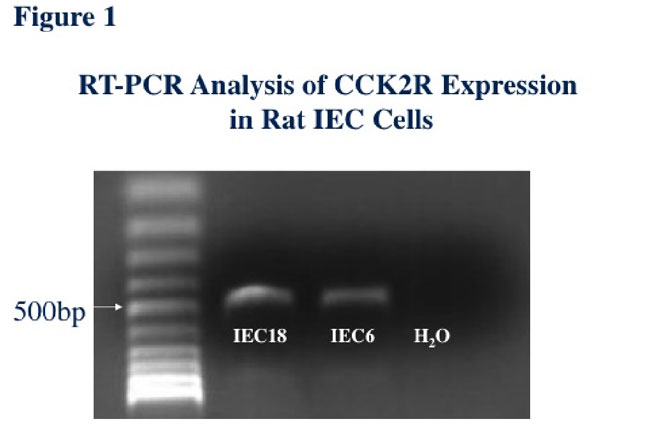
In addition, in unpublished studies, we have identified cell lines that do not express CCK2R, and that upon transfection of CCK2R bind progastrin and show proliferative effects and activated signaling (Jin, G et al., in preparation). As previously published by our group (4), the progastrin interaction with CCK2R does not appear to induce calcium signaling, nor is competition with G-17 sufficient to exclude binding, suggesting interaction with discrete epitopes or a more complicated mechanism. The absence of increased colonic proliferation seen in the INS-GAS mice, which show elevated levels of amidated gastrin, certainly does not discount a role for CCK2R in mediating proliferative effects of progastrin since we believe the interactions to be quite different. Similarly, the fact that glycosaminoglycan proteins can also interact with progastrin would not seem to exclude a specific progastrin-CCK2R interaction.
Dr. Singh has advocated for a pre-eminent role for Annexin-II as a progastrin receptor, and the group has published a number of papers documenting significant interactions between progastrin and Annexin-II. However, as yet there does not appear to be strong genetic evidence, particularly in non-transformed cell types, that these interactions are functionally significant in the whole animal. The expression of Annexin-II appears to be somewhat more ubiquitous and is not highly localized to the crypt cells in the colon that respond to progastrin. Annexin-II does not appear to have the structure of a classical receptor, and while a few studies have suggested expression on the “outer membrane” of epithelial cells, our published studies of IEC-6 cells showed no cell surface staining of Annexin-II in FACS (4). However, we would agree that further work is needed to determine whether progastrin interacts exclusively with CC-2R in vivo or whether there are indeed additional receptors such as Annexin-II that could also play important roles in mediating progastrin’s effects.
Sincerely,
Timothy C. Wang
Guangchung Jin
D. Mark Pritchard
References
- Jin G, Ramanathan V, Quante M, Baik GH, Yang X, Wang SS, Tu S, Gordon SA, Pritchard DM, Varro A, Shulkes A, Wang TC. 2009. Inactivating cholecystokinin-2 receptor inhibits progastrin-dependent colonic crypt fission, proliferation, and colorectal cancer in mice. J Clin Invest. 2009 Sep;119(9):2691–2701.
- Singh P, Velasco M, Given R, Varro A, Wang TC. 2000. Progastrin expression predisposes mice to colon carcinomas and adenomas in response to a chemical carcinogen. Gastroenterology. 119:162–171.
- Singh P, Wu H, Clark C, and Owlia A. 2007. Annexin II binds progastrin and gastrin-like peptides, and mediates growth factor effects of autocrine and exogenous gastrins on colon cancer and intestinal epithelial cells. Oncogene. 26: 425–440.
- Dubeykovskiy A, Nguyen T, Dubeykovskaya Z, Lei S, Wang TC. 2008. Flow cytometric detection of progastrin interaction with gastrointestinal cells. Regul Pept. Nov 29;151(1-3):106–114. Epub 2008 Jul 9.



Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)