Submit a comment
Citation Information: J Clin Invest. 2004;114(10):1457-1466. https://doi.org/10.1172/JCI21982.
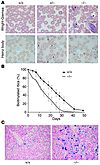
Abstract
Hemoglobin (Hb) A production during red blood cell development is coordinated to minimize the deleterious effects of free α- and β-Hb subunits, which are unstable and cytotoxic. The α-Hb–stabilizing protein (AHSP) is an erythroid protein that specifically binds α-Hb and prevents its precipitation in vitro, which suggests that it may function to limit free α-Hb toxicities in vivo. We investigated this possibility through gene ablation and biochemical studies. AHSP–/– erythrocytes contained hemoglobin precipitates and were short-lived. In hematopoietic tissues, erythroid precursors were elevated in number but exhibited increased apoptosis. Consistent with unstable α-Hb, AHSP–/– erythrocytes contained increased ROS and evidence of oxidative damage. Moreover, purified recombinant AHSP inhibited ROS production by α-Hb in solution. Finally, loss of AHSP worsened the phenotype of β-thalassemia, a common inherited anemia characterized by excess free α-Hb. Together, the data support a model in which AHSP binds α-Hb transiently to stabilize its conformation and render it biochemically inert prior to Hb A assembly. This function is essential for normal erythropoiesis and, to a greater extent, in β-thalassemia. Our findings raise the possibility that altered AHSP expression levels could modulate the severity of β-thalassemia in humans.
Authors
Yi Kong, Suiping Zhou, Anthony J. Kihm, Anne M. Katein, Xiang Yu, David A. Gell, Joel P. Mackay, Kazuhiko Adachi, Linda Foster-Brown, Calvert S. Louden, Andrew J. Gow, Mitchell J. Weiss
Guidelines
The Editorial Board will only consider comments that are deemed relevant and of interest to readers. The Journal will not post data that have not been subjected to peer review; or a comment that is essentially a reiteration of another comment.
- Comments appear on the Journal’s website and are linked from the original article’s web page.
- Authors are notified by email if their comments are posted.
- The Journal reserves the right to edit comments for length and clarity.
- No appeals will be considered.
- Comments are not indexed in PubMed.
Specific requirements
- Maximum length, 400 words
- Entered as plain text or HTML
- Author’s name and email address, to be posted with the comment
- Declaration of all potential conflicts of interest (even if these are not ultimately posted); see the Journal’s conflict-of-interest policy
- Comments may not include figures



Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)