Advertisement
Outcomes of acute leukemia patients transplanted with naive T cell–depleted stem cell grafts
Marie Bleakley,1,2 Shelly Heimfeld,1 Keith R. Loeb,1,3 Lori A. Jones,4 Colette Chaney,1 Stuart Seropian,5 Ted A. Gooley,1 Franziska Sommermeyer,1 Stanley R. Riddell,1,6,7 and Warren D. Shlomchik5,8
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Bleakley, M. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Heimfeld, S. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Loeb, K. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Jones, L. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Chaney, C. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Seropian, S. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Gooley, T. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Sommermeyer, F. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Riddell, S. in: PubMed | Google Scholar
1Fred Hutchinson Cancer Research Center (FHCRC), Seattle, Washington, USA.
2Department of Pediatrics and
3Department of Pathology, University of Washington, Seattle, Washington, USA.
4Seattle Cancer Care Alliance, Seattle, Washington, USA.
5Yale University School of Medicine (YUSM), New Haven, Connecticut, USA.
6Department of Medicine, University of Washington, Seattle, Washington, USA.
7Institute for Advanced Study, Technical University of Munich, Munich, Germany.
8University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Address correspondence to: Marie Bleakley, Program in Immunology, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, Washington 98109, USA. Phone: 206.667.6572; E-mail: mbleakle@fredhutch.org.
Authorship note: Stanley R. Riddell and Warren D. Shlomchik are co–senior authors.
Find articles by Shlomchik, W. in: PubMed | Google Scholar
Published June 8, 2015 - More info
J Clin Invest. 2015;125(7):2677–2689. https://doi.org/10.1172/JCI81229.
Copyright © 2015, American Society for Clinical Investigation
Received: February 4, 2015; Accepted: April 30, 2015
-
Results
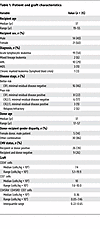
Patient characteristics and graft engineering. The patient characteristics are listed in Table 1. Of 108 patients aged 14 to 55 years with an HLA-MRD (>13 years old) who were evaluated for HCT for the treatment of acute leukemia or advanced MDS during the study period (December 2009 to May 2014), 35 were allocated to HCT in the TN depletion clinical trial and treated accordingly (Figure 1). The major reasons that other patients were not allocated to the trial included allocation to a non-TBI–containing myeloablative HCT at the discretion of the transplant attending physician (in most cases, patients with AML or MDS with better-risk disease and/or comorbidities); allocation to a nonmyeloablative regimen; or allocation to transplantation on a standard myeloablative treatment plan with cyclophosphamide and TBI conditioning. Patients allocated to cyclophosphamide and TBI included patients that lacked insurance coverage for a clinical trial, were unwilling to consent to the trial of TN depletion, or were precluded for participating for logistical reasons or because they did not meet eligibility criteria for the trial.
 Figure 1
Figure 1Consort diagram of phase II nonrandomized clinical trial. Number of patients assessed for eligibility and excluded or allocated to the trial, treated, followed, and analyzed. Patients with AML, ALL, or MDS could be allocated to nonmyeloablative HCT or to less intensive, non-TBI myeloablative HCT if the attending physician determined that their disease status required less intensive conditioning or if the patient had significant co-morbidities. Patients who were allocated to standard HCT with cyclophosphamide TBI conditioning, rather than the TN depletion trial, included patients who refused consent or lacked insurance coverage to participate in a clinical trial, those who were precluded from participating for logistical reasons, and those who did not meet eligibility criteria.
To ensure consistency in the trial, we established target ranges for the CD34+ cells, CD3+CD45RO–CD45RA+ TN, and total CD3+ T cells in the engineered stem cell grafts. A CD34+ cell dose of >5.0 × 106 cells/kg was selected because exceeding this threshold is associated with improved overall survival in recipients of PBSC HCT (31). A target of ≤7.5 × 104 TN/kg was chosen based on estimates that a quantity exceeding this number would be sufficient to cause GVHD. A target of 1 × 107 CD3+ T cells/kg, with an acceptable range of 1 × 106 to 10 × 106 CD3+ T cells/kg, was selected because this range of T cells is 10- to 100-fold greater than the number of unselected T cells predicted to cause GVHD after MRD HCT, and we reasoned that this number was likely to provide sufficient TM to facilitate immune reconstitution (32). We were successful in achieving these targets for all patients. As shown in Table 1, the PBSC grafts administered contained a median of 7.4 × 106 CD34+ cells/kg (range 5.1 × 106 to 19.9 × 106 CD34+ cells/kg) and 107 CD3+ T cells/kg that included 3,600 TN/kg (range 500–74,600 TN/kg; interquartile range 2,200–6,500 TN/kg). Eighty-six percent of patients received <10,000 TN/kg.
Donor cell engraftment. Successful and sustained engraftment of donor hematopoietic cells was a primary safety endpoint of the study. All patients had neutrophil engraftment, achieved at a median of 13 days (range 9–17 days), and platelet counts exceeded 20,000 per mm3 at a median of 14 days (range 9–111 days). Donor and recipient chimerism was determined by DNA genotyping for short tandem repeat polymorphisms. Myeloid (CD33+) engraftment was 100% donor in all recipients, and CD3+ T cells were 100% donor in most recipients after day 28 (Figure 2). There were no graft rejections, although one patient developed secondary graft failure at day 260 while still having 100% donor cells.
 Figure 2
Figure 2Engraftment and chimerism. Cumulative incidence of (A) neutrophil and (B) platelet engraftment. Percentage donor chimerism in sorted (C) CD33+ myeloid cells and (D) CD3+ T cells.
aGVHD. Twenty-three of the thirty-five patients developed clinical symptoms and signs consistent with grades II–IV aGVHD (66%; 95% CI 41%–74%), and the diagnosis was confirmed in all cases by biopsy of an involved site (Figure 3A). A test of the null hypothesis that the true rate of grades II–IV aGVHD is 60% yields P = 1.0 (1-sided binomial test), and therefore, we could not reject our prespecified null hypothesis for the second primary endpoint of the study. Three patients (9%; 95% CI 0%–18%) developed grade III aGVHD, and no cases of grade IV aGVHD or liver aGVHD were observed. The clinical pattern and stage of gastrointestinal (GI) and skin aGVHD are shown in Figure 3, B–D. Ten patients (29%) had no aGVHD; two (6%) developed grade I aGVHD (skin stage 2 only); thirteen (37%) had grade II GVHD limited to GI stage 1; seven (20%) had GI stage 1 and skin stage 1–2; and three (9%) had grade III aGVHD manifested by diarrhea (GI stage 2 [n = 1]; stage 3 [n = 2]) (Figure 3B).
 Figure 3
Figure 3GVHD. (A) Cumulative incidences of aGVHD by grade. (B) aGVHD organ involvement. Clinical severity of (C) GI aGVHD and (D) skin aGVHD. (E) Histopathological severity of GI GVHD on endoscopic biopsy samples. (F) Incidence of cGVHD.
All 23 patients with GI aGVHD developed symptoms while receiving prophylactic tacrolimus. Histologic grading of GVHD severity was performed in each case, and the scores were minimal in 9 patients and mild in 14 patients (Figure 3E). Twenty-two patients received systemic corticosteroids (median initial dose of 1 mg/kg; range 0.5–2 mg/kg) in addition to tacrolimus for treatment of grade II–IV aGVHD. Twenty patients had complete resolution of symptoms in <7 days, and the remaining 3 patients achieved a complete response in 7 to 14 days. Importantly, no patient required a second-line agent for treatment of aGVHD.
cGVHD. Three of the thirty-five TN-depleted recipients developed cGVHD, with a median follow-up of 932 days (range 209–1,826 days), for an estimated probability of 9% at 2 years (95% CI 0%–19%; Figure 3F). One patient had a skin rash and dry eyes (mild cGVHD); one patient had late upper GI GVHD, with mild oral changes (moderate cGVHD); and the third patient had upper GI GVHD, mild dry eyes, and developed deep sclerosis of the skin on the lower extremities during the taper of immunosuppression (severe cGVHD). The cGVHD in the latter patient resolved rapidly after initiation of rituximab, and systemic corticosteroids and rituximab were subsequently discontinued without cGVHD recurrence. All of the patients diagnosed with cGVHD had a preceding history of aGVHD. None of the patients diagnosed with grade III aGVHD subsequently developed cGVHD.
Adverse events and nonrelapse mortality. Adverse events were as expected for patients undergoing HCT following a myeloablative preparative regimen containing TBI (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI81229DS1). Nonrelapse mortality (NRM) occurred in 1 patient due to organ failure before day 100 and in 2 patients due to bacterial infections after day 100, resulting in 1- and 2-year estimates of 9% (95% CI 0%–19%; Figure 4A). No patient younger than 46 years old experienced NRM.
 Figure 4
Figure 4Survival, DFS, relapse, and NRM. The probabilities of (A) NRM, (B) overall survival and DFS, (C) relapse among all patients, and (D) relapse among patients in CR1, without minimal residual disease, or in CR2, CR3, and/or with residual disease.
Overall survival, disease-free survival, and relapse. Seven patients have died, and the 1- and 2-year estimates of overall survival were 82% (95% CI 65%–92%) and 78% (95% CI 59%–89%), respectively, with a median follow-up for the 28 survivors of 932 days (range 209–1,826 days). The disease-free survival (DFS) estimates at 1 and 2 years were 77% (95% CI 59%–88%) and 70% (95% CI 51%–83%), respectively (Figure 4B). The estimated probability of relapse was 14% (95% CI 3%–26%) at 1 year and 21% (95% CI 7%–35%) at 2 years (Figure 4C). Patients transplanted in first complete remission (CR1), without minimal residual disease (“better risk”) or with more advanced disease (beyond CR1 and/or with minimal residual disease; “poor risk”), had cumulative relapse incidences of 13% and 28% at 2 years, respectively (Figure 4D).
Infections. All documented infections occurring in the first 100 days were captured (Supplemental Table 2). EBV reactivation in blood was monitored weekly by PCR for the first 100 days in all patients, and we observed only a single low-level positive EBV PCR test result (41 copies/ml) in one patient. Seven patients developed mild self-resolving BK virus cystitis. CMV reactivation occurred in 19 patients, 54% of the total cohort and 73% of patients at risk based on recipient and/or donor CMV seropositivity.
Immune reconstitution. The recovery of blood lymphocyte subsets is shown in Figure 5. The median numbers of CD8+CD3+ T cells and CD4+CD3+ T cells were 177 per μl and 109 per μl at day 28, numbers which are comparable to those reported after T cell–replete MRD HCT and substantially higher than those reported after TCD HCT (Supplemental Table 3 and ref. 33). CD45RA+CD8+ and CD4+ TN were rarely observed before day 180, and Tregs remained infrequent throughout the first year after HCT (Figure 5). Concurrent with the appearance of TN, both TCR excision circles (TRECs) and TCR diversity, as measured by Vβ spectratyping, increased after day 180 (Figure 5H and Supplemental Figure 1).
 Figure 5
Figure 5Quantitative immune reconstitution. (A–G) Absolute numbers of (A) CD8+CD3+ T cells, (B) CD4+CD3+ T cells, (C) CD56+CD16+CD3– NK cells, (D) CD19+ B cells, (E) CD8+ TN, (F) CD4+ TN, and (G) Tregs. Error bars indicate the median value and interquartile range. The gray shading shows the normal range for each respective subset. H shows numbers of signal-joint TRECs (sjTRECs) per 106 PBMCs.
The recovery of T cells in the blood occurred well before emergence of thymic emigrants, consistent with a contribution from TM administered with the graft. We evaluated the transfer of virus-specific TM in a subset of 7 HLA-A*0201 CMV-seropositive TN-depleted HCT recipients with CMV-seropositive donors by measuring T cells in donor and recipient blood that were specific for a CMV pp65 peptide (pp65NLV) using an HLA A*0201/pp65NLV tetramer. By tetramer staining, T cells specific for pp65NLV were present in donor blood and in the blood of all 7 recipients 28 days after HCT (Figure 6, A and B). The pp65NLV-specific T cells contained CD27+CD28+ TCM and CD28– and/or CD27– TEM subsets and were functional, as demonstrated by an increase in cell frequency and number in temporal association with CMV reactivation and production of IFN-γ and IL-2 after pp65NLV peptide stimulation (Figure 6, A and C).
 Figure 6
Figure 6Transfer of CMV-specific T cells with TN-depleted HCT. (A and B) pp65NLV-specific T cells were detected by MHCI-tetramer staining of peripheral blood samples from HLA-A*0201+ CMV+ donors and in their respective recipients after HCT. (A) The time course of expansion of pp65NLV tetramer+ cells (right y axis, solid lines, absolute number of CD8+ pp65NLV+ T cells/μl in recipient peripheral blood at days 28, 56, and 80 after HCT) in relation to blood CMV copy number (left y axis, dashed lines) for 3 representative patients. Data shown at time 0 (black triangles) are from donor blood (D) (CD8+ pp65NLV+ T cells/μl). The 3 patients shown in A are representative of a total of 7 patients; the data for all 7 are shown in B. (B) The percentage of pp65NLV tetramer+ cells among CD8+ T cells and absolute numbers of pp65NLV tetramer+ CD8+ T cells in peripheral blood. (C) Expression of CD27 and CD28 on pp65NLV tetramer+ cells (blue overlay) and total CD8+ cells (red underlay) from 3 representative patients at day 56 and IL-2 and IFN-γ production on day 56 in response to pp65 peptide stimulation (gated on CD8+ cells).
Analysis of cGVHD in a contemporary T cell–replete cohort. The 9% incidence of cGVHD among TN-depleted HCT recipients is far lower than the rates of 40% to 63% reported for comparable patients transplanted with MRD T cell–replete HCT and ablative conditioning at our center and others (5–7). In a recent retrospective analysis of the frequency of cGVHD fulfilling NIH cGVHD consensus criteria and requiring systemic immunosuppression, a rate of 44% was found for MRD HCT recipients who received PBSCs between 1992 and 2008 (34). In order to exclude the possibility that the rate of cGVHD after MRD HCT had decreased over time independent of our intervention, we compared the incidence of GVHD in recipients of TN-depleted HCT with that in a cohort of patients that received T cell–replete HCT using a standard cyclophosphamide (120 mg/kg) and TBI (12 Gy) conditioning, with calcineurin inhibitor and methotrexate GVHD prophylaxis, during the same time period that our trial was conducted. The TN-depleted and T cell–replete groups were similar in age, disease type, and disease risk (Supplemental Table 4). The two groups did not differ in the frequency of grades II–IV aGVHD, sites of organ involvement (Figure 7A and Supplemental Figure 2, A and B), or aGVHD treatment. Steroid-refractory aGVHD is uncommon in HLA MRD transplantation, and, consistent with this, 0% and 3.1% of TN-depleted and T cell–replete recipients, respectively, developed steroid-refractory GVHD and required a second-line agent. Twenty of twenty-three TN-depleted HCT recipients and sixteen of twenty-two T cell–replete recipients who were treated for aGVHD with steroids responded within 7 days.
 Figure 7
Figure 7GVHD in recipients of TN-depleted PBSC HCT and the contemporary standard myeloablative PBSC HCT cohort. Cumulative incidences of (A) aGVHD and (B) cGVHD. Cumulative incidences of time to discontinuation of (C) systemic corticosteroids and of (D) all immunosuppression in recipients of TN-depleted and T cell–replete PBSC grafts.
Despite the similar incidences of aGVHD in TN-depleted and T cell–replete HCT recipients, cGVHD was far less frequent (9%; CI 0%–19%) after TN-depleted HCT compared with cGVHD after T cell–replete HCT (56%; CI 38%–73%) (Figure 7B). The median time to discontinuation of corticosteroids for GVHD treatment was 85 days in TN-depleted recipients, which was markedly shorter than the median of 853 days for the T cell–replete cohort (Figure 7C). Furthermore, the median time to discontinuation of all immunosuppression was 316 days among TN-depleted recipients and 1,478 days in the T cell–replete cohort (Figure 7D).
Male recipients of female grafts are at greater risk of cGVHD, and the TN-depleted cohort had fewer such recipients than the T cell–replete comparison group. In order to exclude the possibility that this disparity could explain the higher rate of cGVHD in T cell–replete HCT recipients, we analyzed GVHD in gender combinations other than male recipients of female grafts and found that cGVHD occurred in 11% (CI 0%–23%) of TN-depleted recipients and 48% (CI 27%–68%) of recipients of T cell–replete grafts. Thus, the apparent benefit of TN depletion for reducing cGVHD occurred independent of donor/recipient gender.










Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)