Inflammation
Abstract
Emerging data suggest that hypercholesterolemia has stimulatory effects on adaptive immunity and that these effects can promote atherosclerosis and perhaps other inflammatory diseases. However, research in this area has relied primarily on inbred strains of mice, whose adaptive immune system can differ substantially from that of humans. Moreover, the genetically induced hypercholesterolemia in these models typically results in plasma cholesterol levels that are much higher than those in most humans. To overcome these obstacles, we studied human immune system-reconstituted mice (hu-mice) rendered hypercholesterolemic by treatment with AAV8- PCSK9 and a high-fat/high-cholesterol Western-type diet (WD). These mice had a high percentage of human T cells and moderate hypercholesterolemia. Compared with hu-mice having lower plasma cholesterol, the PCSK9-WD mice developed a T cell-mediated inflammatory response in the lung and liver. Human CD4+ and CD8+ T cells bearing an effector memory phenotype were significantly elevated in the blood, spleen, and lungs of PCSK9-WD hu-mice, while splenic and circulating regulatory T cells were reduced. These data show that moderately high plasma cholesterol can disrupt human T cell homeostasis in vivo. This process may not only exacerbate atherosclerosis but also contribute to T cell-mediated inflammatory diseases in the setting of hypercholesterolemia.
Authors
Jonathan D. Proto, Amanda C. Doran, Manikandan Subramanian, Hui Wang, Mingyou Zhang, Erdi Sozen, Christina Rymond, George Kuriakose, Vivette D'Agati, Robert Winchester, Megan Sykes, Yong-Guang Yang, Ira Tabas
Abstract
Recent studies reveal that airway epithelial cells are critical pulmonary circadian pacemaker cells, mediating rhythmic inflammatory responses. Using mouse models, we now identify the rhythmic circadian repressor REV-ERB as essential to the mechanism coupling the pulmonary clock to innate immunity, involving both myeloid, and bronchial epithelial cells in temporal gating and determining amplitude of response to inhaled endotoxin. Dual mutation of REV-ERBα and its paralog REV-ERBβ in bronchial epithelia further augmented inflammatory responses and chemokine activation, but also initiated a basal inflammatory state, revealing a critical homeostatic role for REV-ERB proteins in the suppression of the endogenous pro-inflammatory mechanism in un-challenged cells. However, REV-ERBα plays the dominant role as deletion of REV-ERBβ alone had no impact on inflammatory responses. In turn, inflammatory challenges cause striking changes in stability and degradation of REV-ERBα protein, driven by SUMOylation and ubiquitination. We developed a novel selective oxazole-based inverse agonist of REV-ERB, which protects REV-ERBα protein from degradation and used this to reveal how pro-inflammatory cytokines trigger rapid degradation of REV-ERα in the elaboration of an inflammatory response. Thus, dynamic changes in stability of REV-ERα protein couple the core clock to innate immunity.
Authors
Marie Pariollaud, Julie Gibbs, Thomas Hopwood, Sheila Brown, Nicola Begley, Ryan Vonslow, Toryn Poolman, Baoqiang Guo, Ben Saer, D. Heulyn Jones, James P. Tellam, Stefano Bresciani, Nicholas C.O. Tomkinson, Justyna Wojno-Picon, Anthony W.J. Cooper, Dion A. Daniels, Ryan P. Trump, Daniel Grant, William Zuercher, Timothy M. Willson, Andrew S. MacDonald, Brian Bolognese, Patricia L. Podolin, Yolanda Sanchez, Andrew S.I. Loudon, David W. Ray

Abstract
Obesity is a major risk factor for insulin resistance and type 2 diabetes. In adipose tissue, obesity-mediated insulin resistance correlates with the accumulation of proinflammatory macrophages and inflammation. However, the causal relationship of these events is unclear. Here, we report that obesity-induced insulin resistance in mice precedes macrophage accumulation and inflammation in adipose tissue. Using a mouse model that combines genetically induced, adipose-specific insulin resistance (mTORC2-knockout) and diet-induced obesity, we found that insulin resistance causes local accumulation of proinflammatory macrophages. Mechanistically, insulin resistance in adipocytes results in production of the chemokine monocyte chemoattractant protein 1 (MCP1), which recruits monocytes and activates proinflammatory macrophages. Finally, insulin resistance (high homeostatic model assessment of insulin resistance [HOMA-IR]) correlated with reduced insulin/mTORC2 signaling and elevated MCP1 production in visceral adipose tissue from obese human subjects. Our findings suggest that insulin resistance in adipose tissue leads to inflammation rather than vice versa.
Authors
Mitsugu Shimobayashi, Verena Albert, Bettina Woelnerhanssen, Irina C. Frei, Diana Weissenberger, Anne Christin Meyer-Gerspach, Nicolas Clement, Suzette Moes, Marco Colombi, Jerome A. Meier, Marta M. Swierczynska, Paul Jenö, Christoph Beglinger, Ralph Peterli, Michael N. Hall
Abstract
The immune system is tightly controlled by regulatory processes that allow for the elimination of invading pathogens, while limiting immunopathological damage to the host. In the present study, we found that conditional deletion of the cell surface receptor Toso on B cells unexpectedly resulted in impaired proinflammatory T cell responses, which led to impaired immune protection in an acute viral infection model, while, in a chronic inflammatory context, was associated with reduced immunopathological tissue damage. Toso exhibited its B cell-inherent immunoregulatory function by negatively controlling the pool of IL-10-competent B1 and B2 B cells, which were characterized by a high degree of self-reactivity and were shown to mediate immunosuppressive activity on inflammatory T cell responses in vivo. Our results indicate that Toso is involved in the differentiation/maintenance of regulatory B cells by fine-tuning B cell receptor (BCR)-activation thresholds. Furthermore, we showed that during influenza A-induced pulmonary inflammation the application of Toso-specific antibodies selectively induced IL-10-competent B cells at the site of inflammation and resulted in decreased proinflammatory cytokine production by lung T cells. These findings suggest that Toso may serve as a novel therapeutic target to dampen pathogenic T cell responses via the modulation of IL-10-competent regulatory B cells.
Authors
Jinbo Yu, Vu Huy Hoang Duong, Katrin Westphal, Andreas Westphal, Abdulhadi Suwandi, Guntram A. Grassl, Korbinian Brand, Andrew C. Chan, Niko Föger, Kyeong-Hee Lee
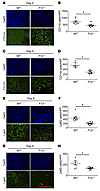
Abstract
Coagulation factor XII (FXII) deficiency is associated with decreased neutrophil migration, but the mechanisms remain uncharacterized. Here, we examine how FXII contributes to the inflammatory response. In 2 models of sterile inflammation, FXII-deficient mice (F12–/–) had fewer neutrophils recruited than WT mice. We discovered that neutrophils produced a pool of FXII that is functionally distinct from hepatic-derived FXII and contributes to neutrophil trafficking at sites of inflammation. FXII signals in neutrophils through urokinase plasminogen activator receptor–mediated (uPAR-mediated) Akt2 phosphorylation at S474 (pAktS474). Downstream of pAkt2S474, FXII stimulation of neutrophils upregulated surface expression of αMβ2 integrin, increased intracellular calcium, and promoted extracellular DNA release. The sum of these activities contributed to neutrophil cell adhesion, migration, and release of neutrophil extracellular traps in a process called NETosis. Decreased neutrophil signaling in F12–/– mice resulted in less inflammation and faster wound healing. Targeting hepatic F12 with siRNA did not affect neutrophil migration, whereas WT BM transplanted into F12–/– hosts was sufficient to correct the neutrophil migration defect in F12–/– mice and restore wound inflammation. Importantly, these activities were a zymogen FXII function and independent of FXIIa and contact activation, highlighting that FXII has a sophisticated role in vivo that has not been previously appreciated.
Authors
Evi X. Stavrou, Chao Fang, Kara L. Bane, Andy T. Long, Clément Naudin, Erdem Kucukal, Agharnan Gandhi, Adina Brett-Morris, Michele M. Mumaw, Sudeh Izadmehr, Alona Merkulova, Cindy C. Reynolds, Omar Alhalabi, Lalitha Nayak, Wen-Mei Yu, Cheng-Kui Qu, Howard J. Meyerson, George R. Dubyak, Umut A. Gurkan, Marvin T. Nieman, Anirban Sen Gupta, Thomas Renné, Alvin H. Schmaier
Abstract
Lupus nephritis (LN) often results in progressive renal dysfunction. The inactive Rhomboid 2 (iRhom2) is a newly identified key regulator of A disintegrin and metalloprotease 17 (ADAM17), whose substrates, such as TNF-α and heparin-binding EGF (HB-EGF), have been implicated in the pathogenesis of chronic kidney disease. Here we demonstrate that deficiency of iRhom2 protects the lupus-prone Fcgr2b–/– mice from developing severe kidney damage without altering anti-double stranded (ds) DNA Ab production, by simultaneously blocking the HB-EGF/EGFR and the TNF-α signaling in the kidney tissues. Unbiased transcriptome profiling of kidneys and kidney macrophages revealed that TNF-α and HB-EGF/EGFR signaling pathways are highly upregulated in Fcgr2b–/– mice; alterations that were diminished in the absence of iRhom2. Pharmacological blockade of either TNF-α or EGFR signaling protected Fcgr2b–/– mice from severe renal damage. Finally, kidneys from LN patients showed increased iRhom2 and HB-EGF expression, with interstitial HB-EGF expression significantly associated with chronicity indices. Our data suggest that activation of iRhom2/ADAM17-dependent TNF-α and EGFR signaling plays a crucial role in mediating irreversible kidney damage in LN, thereby uncovering a novel target for selective and simultaneous dual inhibition of two major pathological pathways in the effector arm of the disease.
Authors
Xiaoping Qing, Yurii Chinenov, Patricia Redecha, Michael Madaio, Joris J.T.H. Roelofs, Gregory Farber, Priya D. Issuree, Laura Donlin, David R. McIlwain, Tak W. Mak, Carl P. Blobel, Jane E. Salmon

Abstract
Macrophages are a source of both proinflammatory and restorative functions in damaged tissue through complex dynamic phenotypic changes. Here, we sought to determine whether monocyte-derived macrophages (MDMs) contribute to recovery after acute sterile brain injury. By profiling the transcriptional dynamics of MDMs in the murine brain after experimental intracerebral hemorrhage (ICH), we found robust phenotypic changes in the infiltrating MDMs over time and demonstrated that MDMs are essential for optimal hematoma clearance and neurological recovery. Next, we identified the mechanism by which the engulfment of erythrocytes with exposed phosphatidylserine directly modulated the phenotype of both murine and human MDMs. In mice, loss of receptor tyrosine kinases AXL and MERTK reduced efferocytosis of eryptotic erythrocytes and hematoma clearance, worsened neurological recovery, exacerbated iron deposition, and decreased alternative activation of macrophages after ICH. Patients with higher circulating soluble AXL had poor 1-year outcomes after ICH onset, suggesting that therapeutically augmenting efferocytosis may improve functional outcomes by both reducing tissue injury and promoting the development of reparative macrophage responses. Thus, our results identify the efferocytosis of eryptotic erythrocytes through AXL/MERTK as a critical mechanism modulating macrophage phenotype and contributing to recovery from ICH.
Authors
Che-Feng Chang, Brittany A. Goods, Michael H. Askenase, Matthew D. Hammond, Stephen C. Renfroe, Arthur F. Steinschneider, Margaret J. Landreneau, Youxi Ai, Hannah E. Beatty, Luís Henrique Angenendt da Costa, Matthias Mack, Kevin N. Sheth, David M. Greer, Anita Huttner, Daniel Coman, Fahmeed Hyder, Sourav Ghosh, Carla V. Rothlin, J. Christopher Love, Lauren H. Sansing

Abstract
The underlying cellular mechanisms of catatonia, an executive “psychomotor” syndrome that is observed across neuropsychiatric diseases, have remained obscure. In humans and mice, reduced expression of the structural myelin protein CNP is associated with catatonic signs in an age-dependent manner, pointing to the involvement of myelin-producing oligodendrocytes. Here, we showed that the underlying cause of catatonic signs is the low-grade inflammation of white matter tracts, which marks a final common pathway in Cnp-deficient and other mutant mice with minor myelin abnormalities. The inhibitor of CSF1 receptor kinase signaling, PLX5622, depleted microglia and alleviated the catatonic symptoms of Cnp mutants. Thus, microglia and low-grade inflammation of myelinated tracts emerged as the trigger of a previously unexplained mental condition. We observed a very high (25%) prevalence of individuals with catatonic signs in a deeply phenotyped schizophrenia sample (n = 1095). Additionally, we found the loss-of-function allele of a myelin-specific gene (CNP rs2070106-AA) associated with catatonia in 2 independent schizophrenia cohorts and also associated with white matter hyperintensities in a general population sample. Since the catatonic syndrome is likely a surrogate marker for other executive function defects, we suggest that microglia-directed therapies may be considered in psychiatric disorders associated with myelin abnormalities.
Authors
Hana Janova, Sahab Arinrad, Evan Balmuth, Marina Mitjans, Johannes Hertel, Mohamad Habes, Robert A. Bittner, Hong Pan, Sandra Goebbels, Martin Begemann, Ulrike C. Gerwig, Sönke Langner, Hauke B. Werner, Sarah Kittel-Schneider, Georg Homuth, Christos Davatzikos, Henry Völzke, Brian L. West, Andreas Reif, Hans Jörgen Grabe, Susann Boretius, Hannelore Ehrenreich, Klaus-Armin Nave

Abstract
During tumor progression, immune system phagocytes continually clear apoptotic cancer cells in a process known as efferocytosis. However, the impact of efferocytosis in metastatic tumor growth is unknown. In this study, we observed that macrophage-driven efferocytosis of prostate cancer cells in vitro induced the expression of proinflammatory cytokines such as CXCL5 by activating Stat3 and NF-κB(p65) signaling. Administration of a dimerizer ligand (AP20187) triggered apoptosis in 2 in vivo syngeneic models of bone tumor growth in which apoptosis-inducible prostate cancer cells were either coimplanted with vertebral bodies, or inoculated in the tibiae of immunocompetent mice. Induction of 2 pulses of apoptosis correlated with increased infiltration of inflammatory cells and accelerated tumor growth in the bone. Apoptosis-induced tumors displayed elevated expression of the proinflammatory cytokine CXCL5. Likewise, CXCL5-deficient mice had reduced tumor progression. Peripheral blood monocytes isolated from patients with bone metastasis of prostate cancer were more efferocytic compared with normal controls, and CXCL5 serum levels were higher in metastatic prostate cancer patients relative to patients with localized prostate cancer or controls. Altogether, these findings suggest that the myeloid phagocytic clearance of apoptotic cancer cells accelerates CXCL5-mediated inflammation and tumor growth in bone, pointing to CXCL5 as a potential target for cancer therapeutics.
Authors
Hernan Roca, Jacqueline D. Jones, Marta C. Purica, Savannah Weidner, Amy J. Koh, Robert Kuo, John E. Wilkinson, Yugang Wang, Stephanie Daignault-Newton, Kenneth J. Pienta, Todd M. Morgan, Evan T. Keller, Jacques E. Nör, Lonnie D. Shea, Laurie K. McCauley

Abstract
The NLRP3 inflammasome is a protein complex responsible for caspase-1–dependent maturation of the proinflammatory cytokines IL-1β and IL-18. Gain-of-function missense mutations in NLRP3 cause the disease spectrum known as the cryopyrin-associated periodic syndromes (CAPS). In this study, we generated Nlrp3-knockin mice on various KO backgrounds including Il1b/Il18-, caspase-1–, caspase-11– (Casp1/11-), and Tnf-deficient strains. The Nlrp3L351P Il1b–/– Il18–/– mutant mice survived and grew normally until adulthood and, at 6 months of age, exhibited marked splenomegaly and leukophilia. Injection of these mice with low-dose LPS resulted in elevated serum TNF levels compared with Nlrp3L351P Casp1/11–/– mice and Il1b–/– Il18–/– littermates. Treatment of Nlrp3A350V mice with the TNF inhibitor etanercept resulted in all pups surviving to adulthood, with normal body and spleen/body weight ratios. Nlrp3A350V Tnf–/– mice showed a similar phenotypic rescue, with marked reductions in serum IL-1β and IL-18, reduced myeloid inflammatory infiltrate in the skin and spleen, and substantial decreases in splenic mRNA expression of both inflammasome components (Nlrp3, Pycard, pro-Casp1) and pro-cytokines (Il1b, Il18). Likewise, we observed a reduction in the expression of both pro-Casp1 and pro-Il1b in cultured Nlrp3A350V Tnf–/– BM-derived DCs. Our data show that TNF is an important transcriptional regulator of NLRP3 inflammasome components in murine inflammasomopathies. Moreover, these results may have therapeutic implications for CAPS patients with partial responses to IL-1–targeted therapies.
Authors
Matthew D. McGeough, Alexander Wree, Maria E. Inzaugarat, Ariela Haimovich, Casey D. Johnson, Carla A. Peña, Raphaela Goldbach-Mansky, Lori Broderick, Ariel E. Feldstein, Hal M. Hoffman
No posts were found with this tag.



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)