Advertisement
Genome-wide hepatitis C virus amino acid covariance networks can predict response to antiviral therapy in humans
Rajeev Aurora,1 Maureen J. Donlin,1,2 Nathan A. Cannon,1 John E. Tavis,1,3 and the Virahep-C Study Group
1Department of Molecular Microbiology and Immunology, 2Department of Biochemistry and Molecular Biology, and 3Saint Louis University Liver Center, Saint Louis University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Rajeev Aurora or John E. Tavis, Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, 1100 South Grand Blvd., St. Louis, Missouri 63104, USA. Phone: (314) 977-8891; Fax: (314) 977-8717; E-mail: aurorar@slu.edu (R. Aurora). Phone: (314) 977-8893; Fax: (314) 977-8717; E-mail: tavisje@slu.edu (J.E. Tavis).
Find articles by Aurora, R. in: JCI | PubMed | Google Scholar
1Department of Molecular Microbiology and Immunology, 2Department of Biochemistry and Molecular Biology, and 3Saint Louis University Liver Center, Saint Louis University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Rajeev Aurora or John E. Tavis, Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, 1100 South Grand Blvd., St. Louis, Missouri 63104, USA. Phone: (314) 977-8891; Fax: (314) 977-8717; E-mail: aurorar@slu.edu (R. Aurora). Phone: (314) 977-8893; Fax: (314) 977-8717; E-mail: tavisje@slu.edu (J.E. Tavis).
Find articles by Donlin, M. in: JCI | PubMed | Google Scholar
1Department of Molecular Microbiology and Immunology, 2Department of Biochemistry and Molecular Biology, and 3Saint Louis University Liver Center, Saint Louis University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Rajeev Aurora or John E. Tavis, Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, 1100 South Grand Blvd., St. Louis, Missouri 63104, USA. Phone: (314) 977-8891; Fax: (314) 977-8717; E-mail: aurorar@slu.edu (R. Aurora). Phone: (314) 977-8893; Fax: (314) 977-8717; E-mail: tavisje@slu.edu (J.E. Tavis).
Find articles by Cannon, N. in: JCI | PubMed | Google Scholar
1Department of Molecular Microbiology and Immunology, 2Department of Biochemistry and Molecular Biology, and 3Saint Louis University Liver Center, Saint Louis University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Rajeev Aurora or John E. Tavis, Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, 1100 South Grand Blvd., St. Louis, Missouri 63104, USA. Phone: (314) 977-8891; Fax: (314) 977-8717; E-mail: aurorar@slu.edu (R. Aurora). Phone: (314) 977-8893; Fax: (314) 977-8717; E-mail: tavisje@slu.edu (J.E. Tavis).
Find articles by Tavis, J. in: JCI | PubMed | Google Scholar
1Department of Molecular Microbiology and Immunology, 2Department of Biochemistry and Molecular Biology, and 3Saint Louis University Liver Center, Saint Louis University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Rajeev Aurora or John E. Tavis, Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, 1100 South Grand Blvd., St. Louis, Missouri 63104, USA. Phone: (314) 977-8891; Fax: (314) 977-8717; E-mail: aurorar@slu.edu (R. Aurora). Phone: (314) 977-8893; Fax: (314) 977-8717; E-mail: tavisje@slu.edu (J.E. Tavis).
Published December 22, 2008 - More info
J Clin Invest. 2009;119(1):225–236. https://doi.org/10.1172/JCI37085.
© 2008 The American Society for Clinical Investigation
Received: August 8, 2008; Accepted: October 22, 2008
Related article:
Abstract
Current treatment for chronic hepatitis C is expensive, is often accompanied by burdensome side effects, and, sadly, fails in almost half of cases. The ability to predict such failures prior to treatment could save a great deal of pain and expense for the patient with HCV. In this issue of the JCI, Aurora and colleagues describe the development of genetic markers predictive of treatment response based on a study of viral sequence variation (see the related article beginning on page 225). Genome-wide covariation analyses of pretreatment virus sequences from 94 patients showed distinct patterns of mutations strongly associated with the ultimate success or failure of treatment. Such analyses suggest markers predictive of response to therapy and may lead to new insights into the underlying biology of hepatitis C.
Authors
Thomas S. Oh, Charles M. Rice
-
Introduction
HCV chronically infects about 3.8 million Americans and causes 8,000–10,000 deaths each year in the United States by inducing liver failure or hepatocellular carcinoma (1). HCV infection is treated with a combination of pegylated IFN-α and ribavirin. Treatment for 24–48 weeks clears the virus — referred to herein as sustained viral response (SVR) — in 50%–60% of genotype 1 patients (2, 3). IFN-α provides the primary antiviral effect and can clear HCV when used alone (4, 5). When ribavirin is taken with IFN-α, it roughly doubles the clearance rate (4–6). There are no effective therapies for patients who fail to clear virus following IFN-α plus ribavirin therapy, and the reasons for the high rate of therapeutic failures are unknown.
HCV is a hepatotropic Flavivirus that persistently infects hepatocytes and some lymphocytes (reviewed in ref. 7). The virus is composed of a lipid envelope derived from host cell membranes in which the viral glycoproteins E1 and E2 are embedded. Within the envelope is a capsid formed by the viral core protein that surrounds the viral RNA genome. The approximately 9,600-nt positive-polarity RNA genome is translated to produce a polyprotein of about 3,010 amino acids (Figure 1). The polyprotein is cleaved to produce 10 viral proteins. The nonstructural proteins, P7 through NS5B, replicate the viral RNA on modified host membranes. After genomic replication, virions assemble and are secreted from the cell noncytolytically. It is believed that all HCV proteins form multiprotein complexes during viral replication and/or during assembly of the virion (8, 9). Furthermore, many HCV proteins interact with cellular proteins to modify host cell responses, particularly the type 1 IFN response that is induced by viral replication and whose role is to suppress viral persistence and spread (10).
 Figure 1
Figure 1The HCV ORF. The approximately 9,600-nt positive-polarity HCV RNA genome encodes a long ORF. The ORF is translated into an approximately 3,010–amino acid polyprotein that is cleaved to 10 mature proteins. The known or predicted functions of the proteins are indicated.
The HCV genome is highly variable, with 6 genotypes that are less than 72% identical at the nucleotide level (11–14). Within the genotypes, subtypes with nucleotide identities of 75%–86% may occur. Individual isolates of a given subtype typically differ by about 8%–10%, and, as HCV replicates as quasispecies, multiple variants differing by up to a few percent exist even within individual patients. Viral genetic variation at the genotype level clearly affects the outcome of antiviral therapy, because genotype 1 is cleared by IFN-α and ribavirin only about half of the time, whereas response rates for genotypes 2 and 3 are typically greater than 80% (3, 15). However, the effect of HCV sequence variation within the major genotypes on response to therapy is not understood.
The Viral Resistance to Antiviral Therapy of Chronic Hepatitis C (Virahep-C) clinical study previously investigated the efficacy of pegylated IFN-α plus ribavirin for treating genotype 1 HCV (16). As part of the Virahep-C study, we analyzed viral genetic patterns associated with response or failure of therapy (17, 18). We sequenced the complete pretreatment HCV open reading frame (ORF) from 94 patients and found that HCV genetic variability among sequences from patients in whom therapy efficiently suppressed the virus was significantly higher than among sequences from patients in whom suppression was minimal. We interpreted the association of higher interpatient HCV genetic diversity with response to therapy to imply that HCV survived in the nonresponders because there were only a few ways to optimize activity of the viral proteins, but many ways to interfere with their function.
Our previous analysis of the Virahep-C sequences treated variation at each amino acid position as being independent from all others. However, this is an oversimplification because amino acids interact with other residues, both within a given protein and in other proteins. Hence, we hypothesized that a global analysis of the viral genome in the context of response to therapy would provide new insights on how the virus may evade the selective pressures applied by the drugs.
To test this hypothesis, we analyzed the complete protein coding region of the pretherapy Virahep-C HCV sequences for pairs of amino acid positions that varied in concert among independent viral isolates (i.e., covariance) and then asked whether there were differences in the patterns of these covariances in responders and nonresponders to therapy. Covariance analysis has previously been used to identify residue pairs directly interacting in 3-dimensional structures (19–24), to infer protein/protein interactions (25), to examine allosteric interactions within proteins (26, 27), and to evaluate the relative functional importance of residues in proteins (28). Our genome-wide analysis revealed the existence of genetic interactions, which we believe to be novel, that were interwoven through the HCV genome. Importantly, these interactions were very different in responders and nonresponders to IFN-based therapy; hence, they may permit prediction of the outcome of therapy.

-
Results
Virahep-C cohort and HCV sequences. The consensus pretreatment sequences for the full-length HCV ORF were previously determined from 94 participants in the Virahep-C study (17). The characteristics of these patients are shown in Table 1. Most analyses were performed on all 94 sequences, stratified either by genotype (1a versus 1b) or by genotype plus outcome of therapy. In most cases, we also stratified the samples by genotype plus the extremes of early (day 28) response to therapy, in order to eliminate confounding nonbiological effects on changes in viral titre (such as insufficient drug intake). The day-28 response categories were Marked, Intermediate, and Poor (see Methods). The day-28 stratifications used the 63 Marked and Poor sequences. Table 2 shows the number of samples in the day-28 and treatment outcome classes for genotypes 1a and 1b.
Identification of covarying amino acid positions. To identify covarying amino acid positions in the HCV ORF, we first created 10 multiple sequence alignments of the Virahep-C sequences: 1a All, 1a Marked, 1a Poor, 1a SVR, 1a Non-SVR, 1b All, 1b Marked, 1b Poor, 1b SVR, and 1b Non-SVR. A covariance score for every possible pair of the 2,955 positions in each alignment was calculated by squaring the difference between the number of observed and expected amino acid pairs and normalizing this difference by the number of entries (excluding gaps) in each column, the observed minus expected squared (OMES) method (29). The null model in this method is the expected number of covarying pairs, based on the independent count of the amino acids at each of the 2 positions. Covarying positions were defined as those pairs with scores of at least 0.5, corresponding to a difference of at least 3 covarying pairs between the observed and expected in an alignment of 16 samples. This cutoff was chosen because it was the lowest value that was not greatly influenced by noise, and it allowed us to retain the maximum amount of potentially informative data (see Methods). The covarying positions and amino acid pairs are shown in Supplemental Table 1 (supplemental material available online with this article; doi: 10.1172/JCI37085DS1).
There were 246 covarying positions in genotype 1a and 280 in genotype 1b (Table 2), representing about 10% of the 2,955 columns in the alignment for each genotype. The covarying positions were spread throughout the genome, with positions in each of the 10 viral genes covarying with positions in each of the other 9 genes. These covarying positions, with the exception of position 1,583, are different than the adaptive mutations required for efficient HCV RNA replication in the replicon culture system (30–32). Furthermore, there was a pronounced underrepresentation among the covariances of residues that have been demonstrated through molecular or biochemical analyses to be essential for protein function. This is because covariance by definition requires genetic variation; thus, very highly conserved positions were excluded from this analysis.
The difference between the number of covarying positions in the Marked and Poor response classes (202 vs. 172 for 1a and 265 vs. 195 for 1b; Table 2) was not significant for either genotype by Fisher’s exact test (P > 0.1). Similarly, no significant differences in the number of covarying positions were found when the SVR and Non-SVR sequences were compared (223 vs. 217 for 1a, 262 vs. 221 for 1b). Therefore, covarying positions were common and widespread in the HCV genome, but there were no significant differences in the number of covarying positions between the response classes.
Covarying positions form networks. Inspection of the covarying positions revealed that one member of a covarying pair often covaried with one or more other positions. This led us to hypothesize that the covarying positions may be linked into a network. To test this hypothesis, we performed a complete clustering analysis on an alignments of all 47 sequences for each genotype, 1a and 1b. The results were presented as graphs, in which the nodes represent positions in the sequence alignment and pairs of nodes were connected by a line (referred to herein as an edge) if they had a covariance score of at least 0.5. We found that the covarying positions indeed formed networks (Figure 2, A and C, and Table 2). Both the networks followed the inverse power law distribution, in which the probability that any node has k edges is given by the equation Pr(k) = k–γ (33, 34). For 1a, γ equaled 0.98 (Figure 2B), and for 1b, γ equaled 1.1 (Figure 2D). This finding indicates that the networks had hub-and-spoke architectures, in which a few nodes covaried with many others, but most nodes covaried with only few others. The covariances (edges) between amino acid positions that were most highly connected (hubs) had scores at or near the maximal value possible for alignments of this size. This indicates that there were very strong selective pressures for certain amino acid combinations and against other combinations at these pairs of positions.
 Figure 2
Figure 2The All covariance networks. Each network is composed of 47 sequences per genotype, 1a (A and B) and 1b (C and D), totaling 94 sequences. (A and C) Networks formed by the covariances. The nodes represent covarying amino acid positions, and the edges represent covariances between the nodes. The most highly connected nodes are in yellow. (B and D) Edge distribution for the genotype networks.
To evaluate the generality of these networks, we sought to determine whether the covariance networks found in the Virahep-C sequences were representative of networks derived from HCV sequences in general circulation. A set of 118 full-length genotype 1a sequences from non–Virahep-C patients was collected, and their polyprotein amino acid sequences were deduced. We randomly chose 10 sets of 47 amino acid sequences from the set of 118, and each set was aligned and independently subjected to covariance analysis. The distribution of the covariance scores in the 10 random sets of 47 sequences were compared in a pairwise manner by the Kolmogorov-Smirnov test, and the P values ranged from 0.23 to 0.38 (P = NS). This indicates that the score distributions were statistically indistinguishable among the 10 random sequence sets. The distribution of covariance scores in the Virahep-C 1a All network (47 sequences) was then compared with the distribution in each of the 10 random permutations. These P values ranged from 0.23 to 0.32. Therefore, the distribution of covariance scores within the Virahep-C data set was very similar to that in non–Virahep-C data sets of equivalent size. In addition, the number of nodes, the number of edges, and the edge density were all similar among the 11 sequence sets, and the top 4 hub nodes were the same for all 11 sets. Therefore, the covariances and covariance networks found in the Virahep-C sequences were representative of a randomly chosen set of HCV sequences.
Connectivity of the covariance networks differs by response class. We next examined the characteristics of the networks that were generated from the sequences when they were stratified by response of the patient to antiviral therapy (Marked, Poor, SVR, or Non-SVR). The characteristics of the 8 response-specific covariance networks are shown in Table 2, and the 1a Marked and 1a Poor networks are shown in Figure 3, A and C. The networks all had a hub-and-spoke architecture, with γ ranging about 1.0 to 1.2, and the numbers of nodes and edges were similar between the contrasting response classes (Marked versus Poor and SVR versus Non-SVR; Table 2). Similarly, more than half of the amino acid positions that formed the nodes in the networks were shared between the contrasting response classes (Figure 4, A and B). However, the pairs of positions forming the networks from the contrasting phenotypes were very different, with relatively few edges shared between them (Table 3 and Figure 4, C and D). This difference in connectivity among the nodes led to differences in the identity of the most highly connected hubs between the networks for the various response classes (Table 4). For example, the positions that were most highly interconnected for 1a were within NS2, NS4A, and E2 in the Marked responders, but in P7, E2, and NS5A in the Poor responders. Furthermore, most nodes within the Poor and Non-SVR nonresponder networks were more highly connected than were most nodes in the Marked and SVR responder networks. An example is in Figure 3, B and D, which compare first-neighbor networks for residue 463 in the 1a Marked and 1a Poor networks. Therefore, networks with similar numbers of covarying amino acid positions were found in all response classes, but the patterns of connections among the covarying residues were very different in the contrasting response classes.
 Figure 3
Figure 3The connectivity of the response-specific covariance networks is different. Shown are 1a Marked (A and B) and 1a Poor (C and D) response classes. (A and C) Networks formed by the covariant pairs of residue positions. The most highly connected nodes are in yellow. (B and D) Positions that directly covary with position 463 (square node) are shown to highlight the differences between the networks.
 Figure 4
Figure 4Segregation of the edges and nodes by phenotype. The overlap and segregation of the covarying nodes (A and B) and edges (C and D) by response class is shown for genotype 1a (A and C) and 1b (B and D).
To test the possibility that the differences between the Marked and Poor networks may have occurred by chance, we generated alignments in which we randomly exchanged sequences between the Marked and Poor responders. The covariance analysis was repeated for 10 iterations at 3 levels of randomly exchanged sequences: 12.5% (2 of 16 genomes), 25% (4 of 16 genomes), and 37.5% (6 of 16 genomes). An example of 1a Marked responders shuffled with 1a Poor responders is shown in Figure 5. Exchanging 2 sequences in the shuffled networks led to the loss of approximately 15% of the nodes and edges (e.g., the residue positions and their interactions) found in the unshuffled alignments. At 4 sequences shuffled, the number of edges and nodes decreased further, and when 6 sequences were shuffled, there was a large loss of the original nodes and edges. In every case, the covarying positions that were present in the shuffled alignments were also found in the All alignment. The results of these control analyses indicate that (a) the response-specific networks are not so sensitive to replacement of sequences that they exist on the edge of chaos; and (b) although the networks are not hypersensitive to mixing of the phenotypic classes, they do depend on the phenotypic clustering of the sequences for their integrity. Together, these observations indicate that the covarying pairs identified for the response classes were not generated by random chance and therefore reflect a feature of the viruses infecting the patients in the various response classes.
 Figure 5
Figure 5Random shuffling of sequences causes loss of information in the response-specific networks. We generated 10 independent alignments, in which 1a Marked sequences were randomly replaced with 1a Poor sequences at 3 levels of replacement: 2, 4, or 6 sequences, giving 12.5%, 25%, and 37.5% sequences shuffled, respectively. Shown are proportions of true positive covarying pairs relative to the unshuffled Marked sequences. P values showing significance of the differences in conserved edges relative to the unshuffled network were determined by Student’s t test.
HCV replicates as a quasispecies, but our analyses were performed with the consensus sequences (the most common residue at each position) from each individual; consequently, the quasispecies variation within an individual could affect the network. The shuffling experiment in Figure 5 revealed that the networks could withstand replacement of at least 25% of the sequences with others that vary by about 10%. The quasispecies variation within an individual is typically approximately 1%, but can reach up to about 4% in some patients; hence, the networks were tolerant of variation that was considerably larger than the quasispecies variability typical for an individual. In addition, analysis of the frequency of each degenerate nucleotide position in the codons for the top 5 hub positions for the Marked and Poor sequences (Table 4) revealed that the probability that a given viral RNA molecule in the quasispecies spectrum encoded all 5 of the hub consensus sequence residues was 93% for the Marked sequences and 82% for the Poor sequences. Finally, near–full-length HCV quasispecies variants have previously been analyzed in 2 genotype 1a patients, with 6 variants sequenced per patient (35, 36). In one patient, the residues at the top 5 hubs from our All network were identical in the consensus sequence derived from the quasispecies variants and in all 6 of the variants, and in the other patient there was a single substitution at one hub position in one of the 6 quasispecies variants. Therefore, the central portion of the All network was perfectly conserved in 11 of the 12 (92%) quasispecies variants for which the integrity of the networks can be assessed.
The spectrum of HCV isolates circulating in the human population appears to range from relatively resistant to relatively sensitive sequences in their responses to IFN-based therapy (17). Therefore, if the covariance networks accurately reflect the HCV population as a whole, essentially every covariance in the All networks — composed of the full set of 47 sequences for each genotype — should be present in at least one of the response-specific networks (i.e., Marked, Poor, SVR, or Non-SVR). To test this prediction, the All networks, generated from alignments of all 47 1a or 1b sequences, were compared with the response-specific networks. As predicted, every node in the All networks was in at least one of the response-specific networks (Figure 4, A and B). Similarly, every edge in the All networks was also found in one or more of the response-specific networks (Figure 4, C and D). This indicates that no new interactions appeared by chance in the full set of sequences and that the information in the All network was also found within the subsets segregated by treatment response.
Topological assessment of the covarying positions. Genetic covariance is indicative of a functional interaction between the covarying residues. Covariance interactions are often caused by direct binding between the residues, but other interactions, such as compensatory allosteric changes, are also common. HCV replication and assembly involve multicomponent complexes composed of viral and possibly cellular proteins. Furthermore, these processes take place in association with cytoplasmic membranes (8, 9); hence, all or nearly all HCV proteins are membrane associated. Therefore, to evaluate whether the covarying residues could interact directly with each other, we evaluated their topological orientation relative to cellular membranes using the experimentally known orientations or inferred orientations for those residues lacking experimental data (8). In 1a, we found that for the 714 covarying amino acid pairs in which both residues were within the same protein, 672 were in the same compartment, 41 were in adjacent compartments (e.g., cytosol and transmembrane), and 1 was in nonadjacent compartments (e.g., lumen and cytosol). For 1b, there were 460 pairs in the same compartment, 38 in adjacent compartments, and 10 in nonadjacent compartments among the 508 covarying pairs in the same protein. When this analysis was expanded to include all interactions in the networks, we found that nearly 75% of the pairs occurred in the same or adjacent compartments for both subtypes. This indicates that many of the covarying pairs could be in direct contact with each other and/or interact with a common partner.
Structural assessment of covarying positions. The possibility that some of the covarying residues may contact each other can be tested for all or part of NS2, NS3, NS5A, and NS5B because crystal structures are available for these proteins. To this end, we mapped the covariant amino acid pairs within these proteins onto the structures. For 1a, there were 39 covarying pairs within the NS2 crystal structure. The covarying residues were on the solvent-accessible surface of the protein for 29 of the pairs, but none of the paired residues were close enough to bind to each other (≤7.5 υ). For 1b, there were 88 pairs in the structure. Of these, 80 were solvent exposed, but the residues in only 1 pair were within 7.5 υ of each other. For NS3, there were 26 covariant pairs for 1a and 32 for 1b. Although all of these pairs were on solvent-exposed surfaces, none of the paired residues were within 7.5 υ of each other. No intraprotein pairs were found within the N-terminal third of NS5A that is in the crystal structure. There were 21 covarying pairs within 1a NS5B and 67 pairs in 1b, and all of these were on the surface of the protein, but again, none of the covarying residues were within 7.5 υ of each other. Therefore, the large majority of the covarying residues in the available protein structures cannot bind directly to each other. However, most of the covarying residues are on the solvent-exposed surfaces of their respective proteins, where they may be involved in intermolecular interactions with other cellular or viral components.
X-ray cocrystal structures are needed to provide sufficient resolution to evaluate the possibility of direct intermolecular binding between covarying residues, and the only such data currently available are for the NS3/NS4A complex (NS4A is a peptide cofactor for NS3 that folds into the NS3 structure). There were 2 covariances in our data set between 1a NS3 and NS4A in which both residues were within the structure, residue pairs 1,106/1,686 and 1,117/1,686. Residues 1,106 (NS3) and 1,686 (NS4A) are separated only by an intervening protein chain; therefore, variations at these sites could easily communicate with each other through minor alterations to the local protein structure. Covarying residues 1,117 (NS3) and 1,686 (NS4A) are located far from each other in 3-dimensional space, and consequently a cascade of allosteric interactions through the protein would be needed for them to communicate with each other. Therefore, the limited data available indicate that the covariance interactions may involve both local and long-distance allosteric interactions.
Nonresponder networks have more hydrophobic pairs. To analyze the nature of the interactions between the covarying residues, we asked whether differences exist in the chemical nature of the covarying amino acid pairs within the networks. In both subtypes, hydrophobic pairs (i.e., Ile-Leu or Phe-Leu) were significantly more common in the Poor responders than in the Marked responders (3.2-fold in 1a and 3.5-fold in 1b; Table 5). Similar excesses of hydrophobic pairs were found in the Non-SVR networks relative to the SVR networks. The high frequency of hydrophobic amino acid pairs was a property of the covarying residue pairs in the networks, and not a feature of the sequences as a whole because there were no significant differences in the average number of hydrophobic amino acids between the Marked and Poor or SVR and Non-SVR sequences in either genotype (Table 5).
 Table 5
Table 5
Number of hydrophobic amino acids in the genome and hydrophobic residue pairs in the covariance networks
Identification of potential biomarkers for prediction of therapy outcome. The differences between the SVR and Non-SVR covariance networks imply that the patterns of covariances in the pretreatment sequences could be used to predict the outcome of therapy. Covariance by definition requires that the amino acid positions being compared are variable in a sequence set; consequently, only a fraction of the sequences will contain any given covarying pair. In our analysis, a given covarying pair was found in at most 60% of the sequences. Therefore, we asked whether subnetworks with a limited number of nodes could be identified that could be used as biomarkers to predict treatment outcome in a majority of patients.
To identify potentially predictive subnetworks, we began with each covariance in the SVR or Non-SVR networks and then added the nearest-neighbor covariances in a stepwise fashion, keeping covariances in the growing subnetworks if they increased the proportion of patients for which the subnetwork was accurately associated with outcome of therapy (i.e., coverage). This process was repeated exhaustively until the coverage for each growing subnetwork could no longer be increased. More than 64,000 subnetworks were created by this process. We then selected the subnetworks that were associated with outcome at 100% accuracy with 0% false positive associations (i.e., the covariances were found in the only the desired outcome class and never in the contrasting class), and then ranked them by their coverage to identify the subnetworks that covered the maximal number of patients. Several hundred subnetworks were found that perfectly associated with outcome and had maximal coverage (95.5%–100%, depending on outcome class). We then limited the results to subnetworks composed solely of covariances with the 10% highest covariance scores to ensure that we were using the most robust regions of the networks. Figure 6 shows 3 examples for each response class. These subnetworks had 4–8 nodes, were 100% accurate, had no false associations, and covered 95.5%–100% of the samples. Therefore, we conclude that patterns of amino acid covariance in a handful of positions are closely associated with the outcome of antiviral therapy, and these covariances are potential biomarkers for prediction of the outcome of therapy.
 Figure 6
Figure 6Example subnetworks associated with outcome of anti-HCV therapy, with 100% accuracy and 0% false coverage rates, that are potential biomarkers for prediction of therapy outcome. The subnetworks contain 1 or more covariances that together are found in the indicated fraction (coverage) of the appropriate response class and are never found in the opposing response class. Polyprotein residue numbers are shown for each subnetwork, and the identity of the mature HCV protein is indicated in subscript.










-
Methods
Patients and response classes. We used 94 viral pretreatment sequences derived from participants in the Virahep-C clinical study (16). Each patient received full doses of pegylated IFN-α and ribavirin for the first 28 days. This research was conducted in accordance with the Helsinki principles: all patients gave informed, written consent prior to their participation in Virahep-C and its associated basic science components, and all components of Virahep-C were approved by their local Institutional Review Boards. The Virahep-C viral genetics cohort (17) was selected to be evenly stratified by genotype, 1a versus 1b, and by day-28 responses to therapy. Marked responders had a decline in HCV titres greater than 3.5 log10 or to undetectable between baseline and day 28 of therapy, Intermediate responders had declines of 3.5–1.4 log10, and Poor responders had declines less than 1.4 log10. In the analyses reported here, the samples were stratified by either genotype (1a or 1b), genotype plus treatment outcome (SVR or Non-SVR), or genotype plus the extremes of early response to therapy (Marked or Poor). The 118 non–Virahep-C HCV genotype 1a sequences used for external validation were downloaded from the Hepatitis C Database of the Broad Institute (http://www.broad.mit.edu/annotation/viral/HCV/Home.html).
Sequencing. Consensus sequences for the HCV ORF were previously obtained by directly sequencing overlapping RT-PCR amplicons as described previously (GenBank accession nos. EF407411–EF407504; refs. 17, 43). Mixed-base positions caused by the HCV quasispecies were resolved by identifying the predominant base at each position. The 3′ end of the HCV ORF could not be amplified in a few samples, hence the C-terminal 56 amino acids of NS5B were excluded from the analyses so that all sequences were represented equally. The numbering system for 1a sequences was identical to the H77 isolate (GenBank accession no. AF009606), and numbering for the 1b sequences was the same as the J4 isolate (GenBank accession no. AF054247).
Calculation of covariance pairs. The sequences were aligned using Clustal W (44) as previously described (17). For each genotype, 5 different alignments were created and independently subjected to covariance analysis (Table 2).
Algorithms that calculate covariance require an intermediate level of conservation. Positions that are invariant do not contain sufficient information to assess correlated variations, and positions that display random differences can generate spurious correlations. Therefore, we evaluated 3 algorithms to identify covarying positions (24, 29, 45). In accord with the results of Fodor and Aldrich (46), we found that the OMES method (29) performed well on this data set. The other methods gave spurious correlations as a result of the relatively high degree of conservation in the HCV sequences or because their clustering methods did not perform well in this context. We used complete clustering with the OMES method because no assumptions (e.g., number or size of the clusters) are needed.
To identify the covarying pairs, we calculated for every possible pair of columns i and j, a score S using observed and expected pairs:
(Equation 1)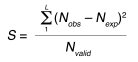
where L is the list of all observed pairs and Nobs is the number of occurrences for a pair of residues. The expected number for the pair (Nexp) is given by:
(Equation 2)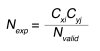
in which Nvalid is the number of sequences in the alignment that are nongap residues, Cxi is the observed number of residues x at position i, and Cyj is the observed number of residues y at position j. The expected number of column pairs calculated in this manner provides a null model for comparisons of the observed pairs.
An OMES score of 0.5 was used as the cutoff for all analyses. This cutoff was chosen for 4 reasons. First, at this value, the All network formed a complete graph (all nodes connected by at least 1 edge), but at a cutoff value of 0.9, 20% of the nodes had no edges. Therefore, 0.5 was not so stringent as to exclude possibly informative data. Second, the number of edges increased dramatically at cutoff values lower than 0.5 (Supplemental Figure 1). A cutoff value of 0.5 corresponded to 3 of 16 possible differences in the pair of columns in the day-28 alignments. [This value is calculated as S = Σ(Nobs – Nexp)2/Nvalid; with S of 0.5 and Nvalid of 16, then 0.5 × 16 = 8 = Σ(Nobs – Nexp)2; the square root of 8 is 2.8, the maximum difference between Nobs and Nexp.] Therefore, covariances with scores less than 0.5 are weak, and using values below 0.5 would greatly increase the number of spurious associations. Third, we observed relatively small effects on the response-specific networks at less than 25% shuffling in the permutation analysis (Figure 4). A cutoff value of 0.5 corresponded to at least 3 of 16 (18.75%) possible differences in the day-28 response–specific alignments, hence this value was well within the stable range of the network. Finally, the number of edges in the Marked and Poor responder networks began to show a difference at a value of 0.5 (Supplemental Figure 1). This was an asset because one of our goals was to find differences between the response classes.
Network generation. Graphs were generated and rendered for the covarying positions in each response class using Cytoscape (version 2.6.1; ref. 47). Such complete clustering is usually not performed on large data sets because it is computationally intensive. However, complete clustering was easily achievable in this case because the covarying positions were obviously linked together. Therefore, we did not need to use the covariance score to generate the clusters. Instead, we simply chose a cutoff value for the score; this is the equivalent of single-linkage clustering.
Statistics. Race and sex distributions were compared across response groups using Pearson’s χ2 test for association. Normally distributed characteristics were compared across response groups using ANOVA, whereas the Kruskal-Wallis nonparametric test was used when distributions were not normally distributed. The fraction of true positives relative to the unshuffled network was compared using a 2-tailed Student’s t test. Fisher’s exact test was used to compare the proportions of hydrophobic pairs between response groups. The program to calculate covariance scores and associated parameters was custom written. Statistical tests were performed using R (version 2.6.2; ref. 48). In all cases, a P value of 0.05 or less was considered significant.
Calculation of solvent exposure. Solvent exposure was calculated using the method of Lee and Richards (49) as described previously (50). The raw exposed surface area was normalized using the tripeptide as a standard state (51). The normalized exposure was averaged over all side-chain atoms, and the residue was considered to be solvent exposed if it had greater than 40% exposed side-chain atoms.
Topology and structural analyses. The transmembrane regions were predicted using TMHMM (52, 53). These regions were then mapped onto the positions of signal sequences and combined with the reported location of proteins (e.g., cytoplasm, endoplasmic reticulum lumen, or transmembrane; refs. 54, 55). Inter-residue distances were calculated using the PDB coordinates (2HD0, 1CU1, 1ZH1, 2GIR, and 2A4Q) using a custom-written program and RASMOL version 2.7 (56).



Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)