Advertisement
Research ArticleImmunologyOncology
Open Access | ![]() 10.1172/JCI183086
10.1172/JCI183086
An activin receptor-like kinase 1–governed monocytic lineage shapes an immunosuppressive landscape in breast cancer metastases
Mehrnaz Safaee Talkhoncheh,1 Jonas Sjölund,1 Paulina Bolivar,1 Ewa Kurzejamska,1,2 Eugenia Cordero,1,3 Teia Vallès Pagès,1 Sara Larsson,1 Sophie Lehn,1 Gustav Frimannsson,1 Viktor Ingesson,1 Sebastian Braun,1 Jessica Pantaleo,1 Clara Oudenaarden,1,4 Martin Lauss,5 R. Scott Pearsall,6 Göran Jönsson,5 Charlotte Rolny,1,7 Matteo Bocci,1,8 and Kristian Pietras1
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Safaee Talkhoncheh, M. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Sjölund, J. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by
Bolivar, P.
in:
PubMed
|
Google Scholar
|

1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Kurzejamska, E. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Cordero, E. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Vallès Pagès, T. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Larsson, S. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Lehn, S. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Frimannsson, G. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Ingesson, V. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Braun, S. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by
Pantaleo, J.
in:
PubMed
|
Google Scholar
|
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Oudenaarden, C. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Lauss, M. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Pearsall, R. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Jönsson, G. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by Rolny, C. in: PubMed | Google Scholar
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by
Bocci, M.
in:
PubMed
|
Google Scholar
|
1Department of Laboratory Medicine, Division of Translational Cancer Research, Lund University Cancer Centre, Medicon Village, Lund University, Lund, Sweden.
2Department of Laboratory Medicine, Karolinska Institutet, Solna, Sweden.
3Lund University Diabetes Centre, Clinical Research Center, Lund University, Lund, Sweden.
4Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark.
5Department of Clinical Sciences, Division of Oncology and Pathology, Lund University Cancer Centre, Lund University, Lund, Sweden.
6P2 Biopharma Consulting, North Reading, Massachusetts, USA.
7Department of Oncology-Pathology, Karolinska Institutet, Solna, Sweden.
8IO Biotech ApS, Copenhagen, Denmark.
Address correspondence to: Kristian Pietras, Lund University Cancer Centre, Division of Translational Cancer Research, Medicon Village, Building 404, 223 81 Lund, Sweden. Phone: 46.709209709; Email: Kristian.Pietras@med.lu.se. Or to: Matteo Bocci, IO Biotech ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark. Phone: 46.707463021; Email: Matteo.Bocci@med.lu.se.
Authorship note: MST and JS contributed equally to this work, and MB and KP contributed equally to this work.
Find articles by
Pietras, K.
in:
PubMed
|
Google Scholar
|
Published January 14, 2025 - More info
J Clin Invest. 2025;135(5):e183086. https://doi.org/10.1172/JCI183086.
© 2025 Talkhoncheh et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: June 7, 2024; Accepted: January 3, 2025

-
Results
Inhibition of ALK1 alters the extent of immune infiltrate in experimental primary and metastatic breast cancer. Based on our previous observations that ACVRL1 expression is associated with immune features of the TME in a series of human solid malignancies (23), we confirmed the generality of this regulation in breast cancer. As expected, the network of genes correlated with ACVRL1 expression paralleled a similar pattern in the The Cancer Genome Atlas (TCGA) Breast Invasive Carcinoma cohort (24, 25), with a highly significant enrichment for angiogenesis and hypoxia, in keeping with the reported endothelial expression of ALK1 (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI183086DS1). However, the largest group of gene set enrichment analysis terms fell into a broad collection of processes defining the functionality and regulation of the immune cell compartment, e.g., IFN-γ response, IL2-STAT5 signaling, and TNF-α signaling via NF-κB (Supplemental Figure 1A and Supplemental Table 1).
In light of these results, we immunostained experimental breast cancer tissue from transgenic MMTV-PyMT mice treated with ALK1-Fc (13) (Figure 1A). Assessment of IHC revealed an increased abundance of immunoreactivity against the broad leukocyte marker CD45 in ALK1-Fc–treated tumors compared with control IgG2a (Figure 1B). An analogous rise was observed for the T lymphocyte–restricted marker CD3 (Figure 1C). Furthermore, RNA was extracted from tissue sections from the same tumors and used as input for a quantitative reverse transcriptase PCR (qRT-PCR) array focusing on immune-related genes. As shown in Figure 1D and Supplemental Table 2, tumors exposed to ALK1-Fc displayed a substantial increase in the expression of genes broadly regulating the identity and activation state of different immune cell types, including Cd40lg, Cd4, Cd3d, Ifng, Il4, and Pdcd1lg2 (PD-L2).
 Figure 1
Figure 1Inhibition of ALK1 alters the extent of immune infiltrate in experimental primary and metastatic breast cancer. (A) Analysis of archival breast cancer tissue from the transgenic MMTV-PyMT mouse model treated with IgG2a or ALK1-Fc (13). (B and C) Representative fields of IHC for CD45 (B), and CD3 (C), ALK1-Fc versus IgG2a (n = 4 for IgG2a, n = 6 for ALK1-Fc). Scale bar: 100 μm. (D) Plot displaying the fold-change expression of target genes from the qRT-PCR, ALK1-Fc versus IgG2a. F.C., fold change. (E) Experimental design of the adjuvant trial based on the orthotopic transplantation of 5 × 105 E0771 cells in syngeneic C57BL/6 hosts (n = 15 for IgG2a, n = 13 for ALK1-Fc). (F) Quantification of macrometastases at sacrifice, ALK1-Fc versus IgG2a. Data are represented as mean with SEM. **P < 0.01, Mann-Whitney U test. (G) Selection of significant gene ontology terms (adjusted P < 0.05) from the analysis performed on bulk RNA-Seq of E0771 lung metastases. Normalized enrichment score (NES) values, ALK1-Fc versus IgG2a. APC, antigen presenting cell.
Primary tumors from MMTV-PyMT mice, or resulting from E0771 breast cancer cell transplantation, did not respond to either programmed cell death protein 1 (PD-1) or cytotoxic T-lymphocyte antigen 4 (CTLA-4) inhibitors alone, or in combination with ALK1-Fc (Supplemental Figure 1, B and C). To better reflect the clinical management of breast cancer patients, and acknowledging the potent immunosuppressive effect of primary tumors (26), we next modeled an adjuvant therapy setup (Figure 1E). Following the engraftment of E0771 breast cancer cells in the abdominal mammary fat pad of syngeneic recipient hosts, incipient tumors were surgically resected when they reached 13 mm in the largest diameter. Five to 7 days after surgery, mice were randomized to receive IgG2a or ALK1-Fc for up to 4 weeks, while being carefully monitored for signs of disease spread to the lungs. At the experimental endpoint, administration of ALK1-Fc significantly reduced the number of macrometastases per mouse, without affecting metastatic incidence (Figure 1F).
Based on the relative size and distribution of the metastases, lung lesions were either manually isolated or laser-capture microdissected, and further processed for bulk RNA-Seq. Enrichment analysis outlined an overrepresentation in tumors from ALK1-Fc–treated mice of pathways related to inflammatory response, complement, and allograft rejection, as well as apoptosis, whereas DNA repair was greatly underscored (Supplemental Figure 1D and Supplemental Table 3). Strikingly, the statistically significant ontology terms covered both the innate and the adaptive arms of the defense response, encompassing more specialized functions such as interleukin production, response to IFN-γ, activation, migration, and chemotaxis of a series of distinct immune cell types (Figure 1G and Supplemental Table 3). Jointly, these data suggest that ALK1 inhibition promotes an inflammatory TME that may be further exploited for improved tumor control.
Murine and human macrophages express Acvrl1/ACVRL1. The exclusive enrichment for immune processes following ALK1 inhibition made us wonder about the etiology of this regulation. Although an altered angiocrine signaling caused by inhibition of ALK1 is a logical and expected possibility considering the reported endothelial specificity of expression, it is tempting to speculate that a population of immune cells may directly react to ALK1 modulation, given the scale of the response observed at the gene expression level. To test the latter hypothesis, primary tumors from MMTV-PyMT mice were dissociated to single-cell suspensions, and expression of Acvrl1 was queried by qRT-PCR of RNA isolated from immune cell populations sorted by flow cytometry (Figure 2A and Supplemental Figure 2A). Isolated endothelial cells and malignant cells served as positive and negative controls for Acvrl1 expression, respectively (Figure 2B). Strikingly, as shown in Figure 2B, a subset of myeloid cells readily expressed Acvrl1, with Ly6C–CD64+ macrophages, CD11b+Ly6Chi monocytes, CD11b+MHCII+ dendritic cells, and CD11b+Ly6G– neutrophils displaying the highest expression levels. Additionally, the expression of Acvrl1 bore functional implications, as stimulation of BM-derived macrophages with the high-affinity ligand BMP9 significantly upregulated the expression of the downstream target gene Id1 (Figure 2C).
 Figure 2
Figure 2Murine and human macrophages express Acvrl1/ACVRL1. (A and B) Study design (A) for the quantification of Acvrl1 expression in FACS-sorted immune cell populations from the MMTV-PyMT model (3 pooled experiments) (B). Positive (CD31+ endothelial cells) and negative (EpCAM+ epithelial cells) controls highlighted in magenta and purple, respectively. Expression of ACVRL1 in freshly isolated human CD14+ monocytes from healthy donors (green). Data are represented as mean with SEM. (C) Expression of Id1 in unstimulated (control) versus BMP9-stimulated BM-derived macrophages (representative of 3 independent experiments). Data are represented as mean with SEM. ***P < 0.001, unpaired, 2-tailed Student’s t test. (D) Overlay of a TAM-specific ACVRL1 signature onto myeloid cells of a human breast cancer scRNA-Seq dataset (32). (E) Expression of ACVRL1 in TAMs from the scRNA-Seq atlas of immune phenotypes (29, 30). Density plot of the expression of ACVRL1 in TAMs. The average expression of the genes composing the TAM signature is presented in the heatmap for ACVRL1+ and ACVRL1– TAM populations. (F) Heatmap of the expression of ACVRL1, cluster markers, and prototypical TAM markers in the scRNA-Seq atlas of immune phenotypes (29). (G) Dual RNAscope ISH coupled with mIHC in human breast cancer. The protein markers CD31 (magenta) and CD45 (white) were used to describe the cellular distribution of the ACVRL1 probe (green). Scale bars: 20 μm; 10 μm (inlet). Two inlets were annotated to highlight endothelial (cyan inlet/arrows) or immune-restricted accumulation of ACVRL1 (yellow inlets/arrows).
Scrutiny of the recently published genomic catalogue of the adult human breast (27) consolidated the expression of ACVRL1 in a series of annotated myeloid cell types (Supplemental Figure 2B), including a variable yet conserved proportion (4%–8%) of macro-M2, macro-lipo, and macro-IFN subsets as the fractions with the highest expression, indicating that ACVRL1 is a feature of several macrophage phenotypes and states. Moreover, our assessment of the expression of ACVRL1 by means of qRT-PCR revealed prominent mRNA expression in freshly isolated human CD14+ monocytes from the peripheral blood of healthy donors (Figure 2B). In agreement with this finding, ACVRL1 was also discernible in CD14+ monocytes in a single-cell RNA-Seq (scRNA-Seq) collection of the human BM (28) (Supplemental Figure 2C).
In the context of cancer, we started off by generating and validating a TAM-specific ACVRL1 signature. Significantly differentially expressed genes (DEGs) between ACVRL1+ and ACVRL1– macrophages were extracted from a breast-specific map of immune phenotypes (29) with an updated cellular annotation (30) (Supplemental Table 4). This list was further filtered through a triple-negative breast cancer (TNBC) scRNA-Seq dataset (31) by applying a stringent criteria selection (i.e., high expression restricted to the macrophage cluster; Supplemental Table 5), leading to 4 genes: SPP1, APOC1, FCER1G, and MMP9. The final signature — which included these 4 genes as well as ACVRL1 — teased out a distinct cluster of myeloid cells and TAMs when imposed on 2 additional breast cancer scRNA-Seq metadata (31, 32) (Supplemental Figure 2, D and E), with the highest average signature expression in lipid-associated macrophages (LAMs) (Figure 2D). Next, we queried the approximately 400 DEGs between ACVRL1+ versus ACVRL1– TAMs in relation to the hallmarks of intratumoral heterogeneity that were recently mapped out (33). Within the macrophage cell type, this analysis confirmed a significant enrichment for lipid-related, glycolysis, and proteasomal degradation metaprograms (Supplemental Figure 2F and Supplemental Table 6).
When closing in on TAMs, inspection of the breast-restricted immune atlas revealed the highest expression of ACVRL1 in angiogenesis-associated, protumorigenic SPP1+ TAMs (34) (Figure 2E and Supplemental Figure 2G). Reassuringly, the average expression of the genes included in the signature was higher in ACVRL1+ versus ACVRL1– TAMs (Figure 2E). Within the SPP1+ cluster, ACVRL1 expression strongly mirrored other immunosuppressive markers, such as FABP5 and TREM2, with both genes further converging into the previously reported LAM phenotype (32, 35) (Figure 2F). To expand on the predicted recruited origin (and supported by ACVRL1 expression in CD14+ monocytes), our analysis highlighted the distinct expression pattern of ACVRL1 relative to FOLR2, which unequivocally identifies resident APOE+ TAMs (36); indeed, FOLR2 expression exclusively segregated in the C1QC+ TAM cluster (Figure 2F).
Finally, to validate these results in patient material, the expression of ALK1 was probed in human breast cancer specimens by RNAscope (due to the paucity of antibodies specific for ALK1 suitable for immunostaining) coupled with highly sensitive multiplexed immunohistochemistry (mIHC) to identify constituent cell types. As expected, the signal of ACVRL1 readily overlapped with the endothelial marker CD31 (cyan inlet/arrows, Figure 2G). Moreover, the characteristic dots of the RNA in situ hybridization (RNA-ISH) clearly accumulated in CD45-positive cells in the tissue (yellow inlet/arrows).
Taken together, our data reveal that expression of ALK1 is not, as previously reported, exclusive for the endothelium and that a population of recruited TAMs is also characterized by ACVRL1 expression across human breast malignancies.
ACVRL1-expressing TAMs display an immunosuppressive phenotype associated with resistance to therapy and poor survival. To gain additional insight into the translational relevance of the molecular cues instigated by ALK1 in macrophages, we estimated survival outcomes in 1,097 breast cancer patients from the TCGA repository (37) through Kaplan-Meier survival fractions, as well as a Cox’s proportional hazard model. After adjusting for stage, age at diagnosis, and estrogen receptor (ER) status, the ACVRL1 signaturehi group exhibited worse disease-specific survival (DSS) (P = 0.017, HR = 1.86 [95% CI = 1.14–3.03]; Figure 3A and Table 1), as well as progression-free interval (PFI) (P = 0.014, HR = 1.66 [95% CI = 116–2.38]; Supplemental Figure 3A), compared with the group of patients with the lowest ACVRL1 signature expression; a similar trend was observed for overall survival (OS) (P = 0.19, HR = 1.23 [95% CI 0.87–1.75]; Supplemental Figure 3B). These results were validated in the METABRIC (38) dataset, which confirmed the significance for DSS (P = 0.018, HR = 1.34, 95% CI= 1.11–1.60; Figure 3B and Table 2), and the trend for OS (P = 0.077, HR = 1.14 [95% CI = 0.99–1.31]; Supplemental Figure 3C).
 Figure 3
Figure 3ACVRL1-expressing TAMs display an immunosuppressive phenotype associated with resistance to therapy and poor survival. (A and B) Survival analysis in the TCGA BRCA (37) (A) and METABRIC (38) (B) datasets. Patients were stratified into 2 risk groups based on the median value of the mean expression of a TAM-specific ACVRL1 signature. The Kaplan-Meier curves show the DSS probabilities of the high (red) and low (green) signature expression groups in the 2 cohorts. P value: log-rank test. The tables summarize the relative Cox’s proportional hazard model analysis for each cohort. (C and D) Box plots depicting the expression of ACVRL1 (C) and the 5-gene signature of ACVRL1+ macrophages (D) in a bulk RNA-Seq dataset of 43 TNBC patients sequenced before treatment with anti–PD-1 (41). Pretreatment features were then correlated to response to therapy (responders, n = 16; nonresponders, n = 27). Statistical analysis was performed using Wilcoxon’s rank sum test, and the P values were corrected for multiple testing with the Benjamini-Hochberg method. (E and F) Expression of ACVRL1 in a CD45+-restricted scRNA-Seq compendium of 48 melanoma patients treated with immune checkpoint inhibitors (40). The average expression of the 5-gene signature, and the average scaled expression of the individual genes are presented in a heatmap (E) based on response, time point, and treatment arm. The average expression of ACVRL1 in the combined CTLA-4 and PD-1 inhibition group was imposed on the UMAP, and further split to create 4 different groups: preresponder, postresponder, prenonresponder, and postnonresponder (F). Contingent on the aggregated data points in F, the average scaled expression of the 5 genes comprised in the ACVRL1 signature is presented in a heatmap (G).
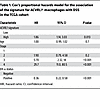 Table 1
Table 1Cox’s proportional hazards model for the association of the signature for ACVRL1+ macrophages with DSS in the TCGA cohort
 Table 2
Table 2Cox’s proportional hazards model for the association of the signature for ACVRL1+ macrophages with DSS in the METABRIC cohort
Prompted by these results, we set out to determine how myeloid expression of ALK1 tied in with the clinical performance of immunotherapy (IT). In a cohort or 43 TNBC patients, in which the pretreatment bulk tumor transcriptional pattern was correlated to the outcome of anti–PD-1 therapy (39), baseline expression of either ACVRL1 alone, or of the gene signature for ACVRL1+ macrophages, was significantly higher in nonresponders (n = 27) versus responders (n = 16) (Figure 3, C and D), indicative of an immunosuppressive environment. However, limited by the evident lack of well-annotated datasets exploring predictive biomarkers for IT in breast cancer and the delay in including IT in standard of care for this malignancy (40), we resorted to melanoma, where the use of immune checkpoint inhibitors has revolutionized the clinical management of patients. Thus, we screened a cohort of 48 melanoma patients that were longitudinally sampled throughout therapy (41), in this case a regimen of anti–CTLA-4, anti–PD-1, or combined CTLA-4 and PD-1 blockade. The analysis of this dataset confirmed that ALK1-expressing macrophages represented a minor subset of TAMs at baseline (circa 7%), which were efficiently removed by therapy in the responder group (Supplemental Figure 3D). Strikingly, ALK1+ TAMs emerged prominently upon treatment resistance, where they represented approximately 18% of the tumor monocyte/macrophage cluster in the nonresponder group (Supplemental Figure 3E). Accordingly, the expression of the ACVRL1 signature — as well as the individual genes included in it — was significantly higher in nonresponders versus responders and in post- versus pretreatment data points (Figure 3E and Supplemental Figure 3E). Notably, the highest frequency and average expression of ACVRL1 were detected in the treatment arm combining dual CTLA-4 and PD-1 blockade. In light of this specific modulation, we homed in on the combined anti–CTLA-4 and anti–PD-1 group, and the data from this treatment arm were dichotomized based on response at each time point. As shown in Figure 3, F and G, the ACVRL1+ myeloid cells present at baseline persisted throughout therapy and further expanded in the nonresponders. This regulation was even more striking when considering the ACVRL1 signature, which showed an inherent difference already at baseline (Supplemental Figure 3E), suggesting that components of this signature might be responsible for intrinsic, primary resistance to IT. Moreover, ACVRL1 expression was tied to that of immunosuppressive markers such as CD274 (PD-L1) and PDCD1LG2 (PD-L2) in nonresponders in the global cohort (Figure 3C). These data imply that ALK1 signaling may be directly involved in the specification of an immunosuppressive phenotype in TAMs. In agreement with this hypothesis, a distinct peak for SMAD5, the signaling mediator downstream of ALK1, could be extrapolated in the promoter region of CD274/PD-L1 from ChIP-Seq data of hematopoietic tissue in the ENCODE database (42, 43), further intersecting with a domain of euchromatin, indicative of direct accessibility to transcription factors (Supplemental Figure 3F). This analysis revealed the specificity of the signal for SMAD5, as the DNA-binding and the histone mark patterns could not be replicated for PDCD1LG2/PD-L2 or HAVCR2/TIM-3 (Supplemental Figure 3, G and H). Collectively, these results offered us a rationale to combine ALK1 blockade with immune checkpoint inhibitors.
Inhibition of ALK1 potentiates IT. Motivated by the previous results indicating an ALK1-dependent specification of a protumorigenic immune state, we yet again exploited our resection-based adjuvant therapy pipeline, this time using the syngeneic 4T1 mammary carcinoma cell line. In an initial trial modeling a premetastatic setting, treatment with ALK1-Fc only showed a trend toward controlling disease progression, possibly reflecting the more aggressive nature of the 4T1 cell line (Supplemental Figure 4, A and B). Next, in a more advanced stage experimental setup, mice were randomized to receive postsurgery therapy with control IgG2a, ALK1-Fc, IT comprising dual PD-1 and CTLA-4 inhibition, or a combination of ALK1-Fc and IT (Figure 4A). At sacrifice, the total lung weight was recorded as a readout for metastatic burden. Notably, the cohort of mice that received combined treatment with ALK1-Fc and IT exhibited the lowest metastatic load of all groups, with a significantly reduced percentage of the lung composed of metastatic lesions, compared with ALK1-Fc alone (Figure 4B).
 Figure 4
Figure 4Inhibition of ALK1 potentiates IT. (A and B) Experimental design of the adjuvant trial based on the orthotopic transplantation of 5 × 104 4T1 cells in syngeneic BALB/c hosts (n = 7 for IgG2a, n = 4 for ALK1-Fc, n = 6 each for IT and ALK1-Fc + IT) (A). IT consists of a dual inhibition of PD-1 and CTLA-4. MFP, mammary fat pad. Quantification of metastatic area in the lungs in the different treated cohorts (B). Data are represented as mean with SEM. *P < 0.05; **P < 0.01; ***P < 0.001, 1-way ANOVA with Bonferroni’s post hoc test for the comparisons between ALK1-Fc versus ALK1-Fc + IT and IT versus ALK1-Fc + IT. (C and D) H&E staining of whole lung sections from the different cohorts in the 4T1 adjuvant trial. Representative pictograms of complete lung metastatic infiltration (green; C) or partial metastatic outgrowth (magenta), quantified in D. Scale bar: 100 μm. P value: χ2 test. (E and F) IHC for CD3 in whole lung sections from the different cohorts (E), and quantification of the proportions of the CD3 distribution in the metastases (F). The staining pattern was arbitrarily categorized as low (cyan), medium (yellow), and high (magenta). Scale bar: 50 μm. P value: χ2 test.
We corroborated these results by assessing the lung parenchyma in the different cohorts. More than 80% of the mice treated with either IgG2a or ALK1-Fc bore metastases that virtually invaded the whole lungs (Figure 4, C and D). Conversely, approximately 60% of the IT group and 85% of the animals in the ALK1-Fc + IT group exhibited distinct malignant lesions that were clearly encapsulated within adjacent/normal lung tissue (Figure 4, C and D). In line with this, evaluation of the distribution of CD3 staining within metastatic lesions supported that control tumors were more generally immune excluded (44, 45) and that they became increasingly more inflamed and readily infiltrated by lymphocytes as IT or a combination of ALK1-Fc + IT were administered (Figure 4, E and F). Even though processing of bulk RNA-Seq did not reveal major transcriptional differences between the cohorts, a series of specific genes with a clear involvement in angiogenesis and immune cell modulation, including Dysf, Ly6g2, Ccl8, and Lrrc32, were significantly up- or downregulated in the combination treatment group versus either monotherapy (Supplemental Figure 4, C–E, and Supplemental Table 7).
Inhibition of ALK1 elicits local and systemic effects on the immune landscape. Next, we sought to define which immune cell type is responsible for the integration of the signals coming from the different inhibitory cues during antiangiogenic IT. For this purpose, resected 4T1 primary tumors, lung metastases, and peripheral blood from a second equivalent experiment were used as input for a multicolor FACS immunophenotyping approach devised to dissect the lymphoid and myeloid arms of the immune system (46) (Figure 5, A and B). Combined treatment with ALK1-Fc and immune checkpoint blockade directly acted on monocytes and macrophages in the metastatic tissue, with a significantly increased frequency of Ly6ChiCD64– and a concomitant reduction of Ly6ChiCD64+ cells (Figure 5, C and D). In line with this, the combined treatment also significantly lowered the proportion of CD64+ macrophages in the lungs (Figure 5E), and increased several subsets of dendritic cells (Figure 5, F–H). In light of the observed expression pattern of ALK1, the combined treatment indirectly altered the lymphoid landscape (Supplemental Figure 5, A–D) by significantly increasing the frequency of NK cells (Figure 5I), CD3+ T cells (Figure 5J), CD4+ T helper cells (Figure 5K), and a trend for CD8+ cytotoxic T lymphocytes (CTLs) (Figure 5L). Moreover, these intrametastatic changes were coupled with systemic alterations in peripheral blood composition (Supplemental Figure 5, E–I), where ALK1-Fc + IT remarkably stalled the differentiation from CD64– to CD64+ monocytes (Figure 5, M–O). This effect appeared to be reliant on ALK1 signaling, as the same modality of regulation was also detected in the ALK1-Fc cohort.
 Figure 5
Figure 5Antiangiogenic IT elicits tumor-specific and systemic effects on the immune cell composition. (A) Experimental design of the 4T1-based adjuvant trial to study different population of myeloid and lymphoid cells via FACS. (B) Representative FACS plots for the myeloid compartment in the different cohorts (n = 5 each for IgG2a and ALK1-Fc, n = 6 each for IT and ALK1-Fc + IT). (C and D) From the CD11b+ gating, relative abundance of monocytes in lung tissue: Ly6ChiCD64– (C), and Ly6ChiCD64+ (D). Data are represented as mean with SEM. P value: unpaired, 2-tailed t test. (E) From the CD11b+ gating, relative abundance of Ly6C– CD64+ macrophages in lung tissue. (F–H) From the CD64– gating, relative frequency of dendritic cells in lung tissue: MHCIIhiCD11Chi (F), MHCIIhiCD11Clo (G), and MHCIIloCD11Chi (G). (I–L) From the CD45+ population, relative frequency of NKP46+ NK cells (I), CD3+ T cells (J), CD4+ T helper cells (K), and CD8+ CTLs (L). (M–O) From the CD45+CD11b+ cells, purity check of circulating monocytes in peripheral blood (M). Relative frequency of circulating monocytes: Ly6ChiCD64– (N) and Ly6ChiCD64+ (O). Data are represented as mean with SEM. *P < 0.05; **P < 0.01; ****P < 0.0001, 1-way ANOVA with Bonferroni’s post hoc test for the comparisons between ALK1-Fc versus ALK1-Fc + IT and IT versus ALK1-Fc + IT.
Altogether, these data suggest a dynamic regulation at multiple levels in the hematopoietic differentiation cascade and the metastatic immune landscape by the addition of ALK1-Fc to an IT regimen.
Vascular immune features reflect differential response to antiangiogenic IT. To combine the data coming from the necropsy observations and the immunophenotyping, we developed a customized mIHC panel to delineate the spatial relationships of endothelial cells and a series of immune cell types (Figure 6A and Supplemental Table 8). In the absence of reliable reagents for the specific detection of ALK1, and to clearly distinguish the presence of different cell types, we selected CD31 to mark endothelial cells and TREM2 as a proxy to identify protumorigenic ALK1+ TAMs. In addition, we stained for tumor infiltrating lymphocytes (TILs): CD4+ T helper cells, CD8a+ CTLs, and B220+ B cells. Finally, (tumor) epithelial cells were distinguished via EpCAM. Following segmentation to classify metastatic tissue and normal/adjacent lung (Figure 6B), cell identification and phenotyping (Figure 6, C and D) mirrored the features captured with FACS, IHC, and RNA-Seq. Given the high level of heterogeneity within and between lesions, we focused on the cohort exposed to combined ALK1-Fc and immune checkpoint blockade, in which we observed a phenotypic range reflecting the overall response surveyed at sacrifice (Figure 6E), as well as for human patients undergoing IT. Metastatic tissue from a mouse that responded to therapy (based on the lung weight and metastatic count at sacrifice, as well as the H&E histology) encompassed the lowest abundance of CD31+ endothelial cells and TREM2+ TAMs (Figure 6E; responder). In contrast, a stable vasculature paralleled by an increased TAM infiltration characterized the lesion of a mouse that had a controlled metastatic burden but relapsed at the primary site (Figure 6E; partial). Finally, prominent metastatic lesion vascularization and a much higher proportion of CD4+ cells within TILs epitomized the group of mice that did not respond/progressed upon combined therapy administration (Figure 6E; nonresponder). This distribution was even more evident when considering the spatial density (Figure 6F): in this analysis, the responder lesion in the combination treatment group displayed the highest density of both T effector and T helper lymphocytes, together with the lowest density of endothelial cells and TAMs. The segmentation further captured that the influx of effector T cells was specific for the metastatic tissue, as the density of the CD8+ CTLs was comparable in the lung segments across the samples. In keeping with this, the CTL-to-macrophage ratio promptly correlated with response (Figure 6G).
 Figure 6
Figure 6Vascular immune features reflect differential response to antiangiogenic IT. (A) Customized multiplexed IHC (mIHC) staining of 4T1 metastases to the lungs. An antibody panel was developed to detect CD31+ endothelial cells (orange), EpCAM+ epithelial cells (both lung epithelium and breast cancer cells, cyan), TILs (CD4+ T helper, green; CD8a+ CTLs, red; B220+ B-cells, yellow), and TREM2+ recruited TAMs (magenta). Scale bars: 50 μm. (B–D) A machine learning-based algorithm was trained to discriminate metastatic tissue (red) from lung and hollow space (green and blue, respectively; B), followed by cell segmentation (C). Phenotyping (D) is visualized as a dot with the same color coding as in B. Cells (based on DAPI detection) negative for any of the markers included in the antibody panel are displayed in blue. (E–G) total cell counts per phenotype (E), and cell density per phenotype (cells/mm2; F) within the different tissue segments. The CD8+ T cells/TREM2+ TAMs ratio (G) was calculated from cell densities.
Taken together, spatial analysis of the cellular composition of responding or resistant lung metastatic lesions revealed that the combination of ALK1-Fc with IT promoted an active antitumor environment, while preserving sensitivity to the antiangiogenic therapy.
ALK1 affects the HPC niche in the BM. Remodeling of the metastatic TME based on the response to ALK1-Fc and IT also corroborated the known and expected ALK1-dependent effects on the vasculature. Thus, we next sought to clarify whether endothelial ALK1 modulation limited the transendothelial migration of myeloid cells, as an explanation for the observed paucity of immunosuppressive TAMs in the metastatic milieu following ALK1 inhibition. To this end, murine lung endothelial cells (mLECs) engineered to express 2 different shRNA constructs against Acvrl1 (shA07 and shA09; Supplemental Figure 6A) or a scrambled control construct (shCtrl) were seeded to populate the top channel of a 3-lane microfluidic organ-on-a-chip device (Figure 7A). A barrier integrity test confirmed the generation of an intact 3D vascular tube, allowing its perfusion with macrophages (Supplemental Figure 6B and Supplemental Video 1). In turn, the CCL2 chemoattractant in the bottom lane stimulated the macrophages to migrate to the intermediate channel. Laser scanning confocal microscopy revealed a consistent trend toward a curtailed migration through Acvrl1-silenced endothelium across our longitudinal imaging schedule (Figure 7B), with a significant difference for the shA09 at 48 hours, indicating that ALK1 indeed regulates extravasation of monocytes.
 Figure 7
Figure 7ALK1 affects the HPC niche in the BM. (A) Experimental design based on the organ-on-a-chip assay with 3 channels. Representative images of a longitudinal and a cross section of the 3D tube are presented in the bottom panel. (B) Quantification of macrophage transendothelial migration at 24 and 48 hours with endothelial cells infected with lentiviral vectors expressing either scrambled or Acvrl1-targeting shRNA (n = 4 experiments). Data are represented as mean with SEM. *P < 0.05, unpaired, 2-tailed Student’s t test for the comparison between shCtrl and shA07 or shA09. (C–E) Experimental design of the short-term trial based on the orthotopic transplantation of 5 × 104 4T1 cells in syngeneic BALB/c hosts (tumor-free: n = 5 each for IgG2a and ALK1-Fc; neoadjuvant: n = 4 each for IgG2a and ALK1-Fc; adjuvant: n = 4 for IgG2a, n = 5 for ALK1-Fc) (C). Frequency of circulating Ly6ChiCD64– (D) and CD64+ (E) monocytes in peripheral blood. Data are represented as mean with SEM. (F) Frequency of Ly6C–CD64+ macrophages in lungs. Data are represented as mean with SEM. **P < 0.01, unpaired, 2-tailed Student’s t test. (G–I) FACS plot and gating strategy of c-Kit+Lin–Sca1– progenitor cells (47) extracted from the BM (G). Frequency of c-Kit+ (H) and GMP (I) cells from the BM extracts. Data are represented as mean with SEM. **P < 0.01, unpaired, 2-tailed Student’s t test. (J) Quantification of colony formation plating efficiency of c-Kit–enriched BM cells. Data are represented as mean with SEM. *P < 0.05, unpaired, 2-tailed Student’s t test. (K) Drawing summarizing the findings.
Subsequently, given the modulation of circulating monocyte populations observed following adjuvant ALK1 inhibition (Figure 5, N and O), we investigated whether ALK1 signaling acted upstream of macrophage functional states. We set up an in vivo experiment based on the 4T1 resection model to monitor the circulating monocytes in the framework of a short-term pharmacological inhibition of ALK1 (Figure 7C). Based on Ly6C and CD64 gating in flow cytometry analysis, we determined that ALK1-Fc reduced the frequency of circulating Ly6ChiCD64+ monocytes in tumor-free mice, and produced a similar trend in the adjuvant setting (Figure 7, D and E, and Supplemental Figure 6C). Similarly, infiltration of CD64+ macrophages was significantly blunted in tumor-free lungs of ALK1-Fc–treated mice compared with the IgG2a group (Figure 7F), indicating that mobilization of monocytes is under the control of ALK1 activity and independent of altered angiogenic stimuli emanating from the tumor.
Taking these results into account, we next sought to clarify which step of the hematopoietic cascade is reliant on ALK1. BM cell evaluation (47) from the experimental groups showed a significant reduction in the population corresponding to the granulocyte-macrophage progenitors (GMP) in the adjuvant ALK1-Fc group, while there was no significant change in the proportion of c-Kit+ progenitor cells (Figure 7, G–I). In agreement with the reduction of BM-GMPs, we also observed a significant decrease in the potential of BM-derived progenitors to differentiate ex vivo into colony-forming unit–granulocyte-macrophage (CFU-GM) colonies following exposure to adjuvant ALK1-Fc treatment in vivo (Figure 7J).
In conclusion, our data demonstrate that ALK1 functionally regulates the microenvironment in 2 specific compartments: locally, by pruning angiogenesis and fine-tuning permissiveness to monocyte/macrophage transendothelial migration; and systemically, by determining the breadth of monocyte differentiation and mobilization in the BM.












Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)