Issue published October 1, 2004 Previous issue | Next issue

- Volume 114, Issue 7
Go to section:

-
Science and Society
×
Abstract
Despite great advances in health-related research and health care, major challenges remain regarding the causes and cures of many diseases; these may be overcome with further research. Our society is enthusiastic about fostering such investigations. However, available federal funds limit many such projects. Previously there have been sizable increases in the NIH budget, but because of the escalating cost of scientific investigation and the pressures of financing other much-needed governmental programs, recent growth in biomedical research funding has barely kept up with inflation. This article focuses on select attempts to sustain the record of scientific achievement enabled in the past by continued increasing investment and also suggests some solutions.
Authors
H. George Mandel, Elliot S. Vesell


-
Commentaries
×
Abstract
Angiogenesis is regulated in large part by the balance of various proangiogenic stimulators, such as VEGF, and a diverse group of endogenous inhibitors of angiogenesis, most of which are extrinsic to endothelial cells. With respect to the latter, until recently, none have appeared to be induced as a consequence of a specific, self-regulating, feedback inhibition response. A new inhibitor, called vasohibin, has been uncovered. Vasohibin is selectively induced in endothelial cells by proangiogenic stimulatory growth factors such as VEGF; it appears to operate as an intrinsic and highly specific feedback inhibitor of activated endothelial cells engaged in the process of angiogenesis.
Authors
Robert S. Kerbel
×Abstract
The etiology of type 2 diabetes is characterized by obesity, insulin and leptin resistance, and compensatory β cell hyperplasia followed by islet degeneration, resulting in the eventual dysregulation of glucose and lipid homeostasis. The recent identification of insulin receptor substrate 2 (IRS2) as a central player in the pathophysiology of many of these processes suggests a potentially unifying molecular link underlying the initiation and progression of type 2 diabetes.
Authors
Matthew J. Brady
×Abstract
Hemoglobinopathies are caused by abnormal structure or synthesis of hemoglobin chains and represent serious monogenic disorders. A new study demonstrates that lentiviral vectors can express clinically relevant levels of human transgenic β-globin in red cells of xenografted mice. While some safety concerns must be addressed, this study is an important step toward potential clinical trials of gene therapy for hemoglobinopathies.
Authors
Christof von Kalle, Christopher Baum, David A. Williams
×Abstract
It has been difficult to develop therapies that target those T cells initiating and mediating the pathogenesis of autoimmune disease. Indeed, most current treatments indiscriminately affect both the autoreactive T cells and the “good” T cells, putting the patient at risk of compromised immune function. A new approach raises the possibility of targeted therapy for autoimmunity. Transplantation of hematopoietic stem cells modified to express a protective form of MHC class II corrects a defect in central tolerance. This method contrasts with other targeted therapies that attempt to modify peripheral tolerance, which is also defective in type 1 diabetes mellitus.
Authors
Rémi J. Creusot, C. Garrison Fathman
×Abstract
The production of protective neutralizing antibodies occurs quickly in some viral infections but very slowly in others. In a new study, surface glycoproteins (the targets of neutralization) of 2 different viruses were genetically switched. Analysis of the neutralizing antibody response to each of the 2 parent and recombinant viruses in infected mice revealed that the speed of neutralizing antibody induction was intrinsically dependent on the surface glycoprotein and not the rest of the virus.
Authors
Eva Szomolanyi-Tsuda, Raymond M. Welsh





-
Research Articles
×
Abstract
Negative feedback is a crucial physiological regulatory mechanism, but no such regulator of angiogenesis has been established. Here we report a novel angiogenesis inhibitor that is induced in endothelial cells (ECs) by angiogenic factors and inhibits angiogenesis in an autocrine manner. We have performed cDNA microarray analysis to survey VEGF-inducible genes in human ECs. We characterized one such gene, KIAA1036, whose function had been uncharacterized. The recombinant protein inhibited migration, proliferation, and network formation by ECs as well as angiogenesis in vivo. This inhibitory effect was selective to ECs, as the protein did not affect the migration of smooth muscle cells or fibroblasts. Specific elimination of the expression of KIAA1036 in ECs restored their responsiveness to a higher concentration of VEGF. The expression of KIAA1036 was selective to ECs, and hypoxia or TNF-α abrogated its inducible expression. As this molecule is preferentially expressed in ECs, we designated it “vasohibin.” Transfection of Lewis lung carcinoma cells with the vasohibin gene did not affect the proliferation of cancer cells in vitro, but did inhibit tumor growth and tumor angiogenesis in vivo. We propose vasohibin to be an endothelium-derived negative feedback regulator of angiogenesis.
Authors
Kazuhide Watanabe, Yasuhiro Hasegawa, Hiroshi Yamashita, Kazue Shimizu, Yuanying Ding, Mayumi Abe, Hideki Ohta, Keiichi Imagawa, Kanji Hojo, Hideo Maki, Hikaru Sonoda, Yasufumi Sato
×Abstract
The molecular link between obesity and β cell failure that causes diabetes is difficult to establish. Here we show that a conditional knockout of insulin receptor substrate 2 (Irs2) in mouse pancreas β cells and parts of the brain — including the hypothalamus —increased appetite, lean and fat body mass, linear growth, and insulin resistance that progressed to diabetes. Diabetes resolved when the mice were between 6 and 10 months of age: functional β cells expressing Irs2 repopulated the pancreas, restoring sufficient β cell function to compensate for insulin resistance in the obese mice. Thus, Irs2 signaling promotes regeneration of adult β cells and central control of nutrient homeostasis, which can prevent obesity and diabetes in mice.
Authors
Xueying Lin, Akiko Taguchi, Sunmin Park, Jake A. Kushner, Fan Li, Yedan Li, Morris F. White
×Abstract
We previously demonstrated that insulin receptor substrate 2 (Irs2) KO mice develop diabetes associated with hepatic insulin resistance, lack of compensatory β cell hyperplasia, and leptin resistance. To more precisely determine the roles of Irs2 in β cells and the hypothalamus, we generated β cell–specific Irs2 KO and hypothalamus-specific Irs2 knockdown (βHT-IRS2) mice. Expression of Irs2 mRNA was reduced by approximately 90% in pancreatic islets and was markedly reduced in the arcuate nucleus of the hypothalamus. By contrast, Irs2 expression in liver, muscle, and adipose tissue of βHT-IRS2 mice was indistinguishable from that of control mice. The βHT-IRS2 mice displayed obesity and leptin resistance. At 4 weeks of age, the βHT-IRS2 mice showed normal insulin sensitivity, but at 8 and 12 weeks, they were insulin resistant with progressive obesity. Despite their normal insulin sensitivity at 8 weeks with caloric restriction, the βHT-IRS2 mice exhibited glucose intolerance and impaired glucose-induced insulin secretion. β Cell mass and β cell proliferation in the βHT-IRS2 mice were reduced significantly at 8 and 12 weeks but not at 10 days. Insulin secretion, normalized by cell number per islet, was significantly increased at high glucose concentrations in the βHT-IRS2 mice. We conclude that, in β cells and the hypothalamus, Irs2 is crucially involved in the regulation of β cell mass and leptin sensitivity.
Authors
Naoto Kubota, Yasuo Terauchi, Kazuyuki Tobe, Wataru Yano, Ryo Suzuki, Kohjiro Ueki, Iseki Takamoto, Hidemi Satoh, Toshiyuki Maki, Tetsuya Kubota, Masao Moroi, Miki Okada-Iwabu, Osamu Ezaki, Ryozo Nagai, Yoichi Ueta, Takashi Kadowaki, Tetsuo Noda
×Abstract
The insulin and IGF signaling pathways are critical for development and maintenance of pancreatic β cell mass and function. The serine-threonine kinase Akt is one of several mediators regulated by these pathways. We have studied the role of Akt in pancreatic β cell physiology by generating transgenic mice expressing a kinase-dead mutant of this enzyme in β cells. Reduction of Akt activity in transgenic animals resulted in impaired glucose tolerance due to defective insulin secretion. The mechanisms involved in dysregulation of secretion in these mice lie at the level of insulin exocytosis and are not the result of abnormalities in glucose signaling or function of voltage-gated Ca2+ channels. Therefore, transgenic mice showed increased susceptibility to developing glucose intolerance and diabetes following fat feeding. These observations suggest that Akt plays a novel and important role in the regulation of distal components of the secretory pathway and that this enzyme represents a therapeutic target for improvement of β cell function in diabetes.
Authors
Ernesto Bernal-Mizrachi, Szabolcs Fatrai, James D. Johnson, Mitsuru Ohsugi, Kenichi Otani, Zhiqiang Han, Kenneth S. Polonsky, M. Alan Permutt
×Abstract
The Raf/MEK/extracellular signal–regulated kinase (ERK) signaling pathway regulates diverse cellular processes such as proliferation, differentiation, and apoptosis and is implicated as an important contributor to the pathogenesis of cardiac hypertrophy and heart failure. To examine the in vivo role of Raf-1 in the heart, we generated cardiac muscle–specific Raf-1–knockout (Raf CKO) mice with Cre-loxP–mediated recombination. The mice demonstrated left ventricular systolic dysfunction and heart dilatation without cardiac hypertrophy or lethality. The Raf CKO mice showed a significant increase in the number of apoptotic cardiomyocytes. The expression level and activation of MEK1/2 or ERK showed no difference, but the kinase activity of apoptosis signal–regulating kinase 1 (ASK1), JNK, or p38 increased significantly compared with that in controls. The ablation of ASK1 rescued heart dysfunction and dilatation as well as cardiac fibrosis. These results indicate that Raf-1 promotes cardiomyocyte survival through a MEK/ERK–independent mechanism.
Authors
Osamu Yamaguchi, Tetsuya Watanabe, Kazuhiko Nishida, Kazunori Kashiwase, Yoshiharu Higuchi, Toshihiro Takeda, Shungo Hikoso, Shinichi Hirotani, Michio Asahi, Masayuki Taniike, Atsuko Nakai, Ikuko Tsujimoto, Yasushi Matsumura, Jun-ichi Miyazaki, Kenneth R. Chien, Atsushi Matsuzawa, Chiharu Sadamitsu, Hidenori Ichijo, Manuela Baccarini, Masatsugu Hori, Kinya Otsu
×Abstract
Phosphorylation of the cell adhesion protein CEACAM1 increases insulin sensitivity and decreases insulin-dependent mitogenesis in vivo. Here we show that CEACAM1 is a substrate of the EGFR and that upon being phosphorylated, CEACAM1 reduces EGFR-mediated growth of transfected Cos-7 and MCF-7 cells in response to EGF. Using transgenic mice overexpressing a phosphorylation-defective CEACAM1 mutant in liver (L-SACC1), we show that the effect of CEACAM1 on EGF-dependent cell proliferation is mediated by its ability to bind to and sequester Shc, thus uncoupling EGFR signaling from the ras/MAPK pathway. In L-SACC1 mice, we also show that impaired CEACAM1 phosphorylation leads to ligand-independent increase of EGFR-mediated cell proliferation. This appears to be secondary to visceral obesity and the metabolic syndrome, with increased levels of output of free fatty acids and heparin-binding EGF-like growth factor from the adipose tissue of the mice. Thus, L-SACC1 mice provide a model for the mechanistic link between increased cell proliferation in states of impaired metabolism and visceral obesity.
Authors
George A. Abou-Rjaily, Sang Jun Lee, Denisa May, Qusai Y. Al-Share, Anthony M. DeAngelis, Randall J. Ruch, Michael Neumaier, Holger Kalthoff, Sue-Hwa Lin, Sonia M. Najjar
×Abstract
Transplantation of genetically corrected autologous hematopoietic stem cells is an attractive approach for the cure of sickle-cell disease and β-thalassemia. Here, we infected human cord blood cells with a self-inactivating lentiviral vector encoding an anti-sickling βA-T87Q-globin transgene and analyzed the transduced progeny produced over a 6-month period after transplantation of the infected cells directly into sublethally irradiated NOD/LtSz-scid/scid mice. Approximately half of the human erythroid and myeloid progenitors regenerated in the mice containing the transgene, and erythroid cells derived in vitro from these in vivo–regenerated cells produced high levels of βA-T87Q-globin protein. Linker-mediated PCR analysis identified multiple transgene-positive clones in all mice analyzed with 2.1 ± 0.1 integrated proviral copies per cell. Genomic sequencing of vector-containing fragments showed that 86% of the proviral inserts had occurred within genes, including several genes implicated in human leukemia. These findings indicate effective transduction of very primitive human cord blood cells with a candidate therapeutic lentiviral vector resulting in the long-term and robust, erythroid-specific production of therapeutically relevant levels of β-globin protein. However, the frequency of proviral integration within genes that regulate hematopoiesis points to a need for additional safety modifications.
Authors
Suzan Imren, Mary E. Fabry, Karen A. Westerman, Robert Pawliuk, Patrick Tang, Patricia M. Rosten, Ronald L. Nagel, Philippe Leboulch, Connie J. Eaves, R. Keith Humphries
×Abstract
The endocrine pancreas undergoes major remodeling during neonatal development when replication of differentiated β cells is the major mechanism by which β cell mass is regulated. The molecular mechanisms that govern the replication of terminally differentiated β cells are unclear. We show that during neonatal development, cyclin D2 expression in the endocrine pancreas coincides with the replication of endocrine cells and a massive increase in islet mass. Using cyclin D2–/– mice, we demonstrate that cyclin D2 is required for the replication of endocrine cells but is expendable for exocrine and ductal cell replication. As a result, 14-day-old cyclin D2–/– mice display dramatically smaller islets and a 4-fold reduction in β cell mass in comparison to their WT littermates. Consistent with these morphological findings, the cyclin D2–/– mice are glucose intolerant. These results suggest that cyclin D2 plays a key role in regulating the transition of β cells from quiescence to replication and may provide a target for the development of therapeutic strategies to induce expansion and/or regeneration of β cells.
Authors
Senta Georgia, Anil Bhushan
×Abstract
The autoimmune disease type 1 diabetes in humans and NOD mice is determined by multiple genetic factors, among the strongest of which is the inheritance of diabetes-permissive MHC class II alleles associated with susceptibility to disease. Here we examined whether expression of MHC class II alleles associated with resistance to disease could be used to prevent the occurrence of diabetes. Expression of diabetes-resistant MHC class II I-Aβ chain molecules in NOD mice following retroviral transduction of autologous bone marrow hematopoietic stem cells prevented the development of autoreactive T cells by intrathymic deletion and protected the mice from the development of insulitis and diabetes. These data suggest that type 1 diabetes could be prevented in individuals expressing MHC alleles associated with susceptibility to disease by restoration of protective MHC class II expression through genetic engineering of hematopoietic stem cells.
Authors
Chaorui Tian, Jessamyn Bagley, Nathalie Cretin, Nilufer Seth, Kai W. Wucherpfennig, John Iacomini
×Abstract
The development of autoimmune diabetes in the nonobese diabetic (NOD) mouse results from a breakdown in tolerance to pancreatic islet antigens. CD28-B7 and CD40 ligand–CD40 (CD40L-CD40) costimulatory pathways affect the development of disease and are promising therapeutic targets. Indeed, it was shown previously that diabetes fails to develop in NOD–B7-2–/– and NOD-CD40L–/– mice. In this study, we examined the relative role of these 2 costimulatory pathways in the balance of autoimmunity versus regulation in NOD mice. We demonstrate that initiation but not effector function of autoreactive T cells was defective in NOD–B7-2–/– mice. Moreover, the residual proliferation of the autoreactive cells was effectively controlled by CD28-dependent CD4+CD25+ regulatory T cells (Treg’s), as depletion of Treg’s partially restored proliferation of autoreactive T cells and resulted in diabetes in an adoptive-transfer model. Similarly, disruption of the CD28-B7 pathway and subsequent Treg deletion restored autoimmunity in NOD-CD40L–/– mice. These results demonstrate that development of diabetes is dependent on a balance of pathogenic and regulatory T cells that is controlled by costimulatory signals. Thus, elimination of Treg’s results in diabetes even in the absence of costimulation, which suggests a need for alternative strategies for immunotherapeutic approaches.
Authors
Hélène Bour-Jordan, Benoît L. Salomon, Heather L. Thompson, Gregory L. Szot, Matthew R. Bernhard, Jeffrey A. Bluestone
×Abstract
Delayed and weak virus neutralizing antibody (nAb) responses represent a hallmark correlating not only with the establishment of persistent infection but also with unsuccessful vaccine development. Using a reverse genetic approach, we evaluated possible underlying mechanisms in 2 widely studied viral infection models. Swapping the glycoproteins (GPs) of lymphocytic choriomeningitis virus (LCMV, naturally persisting, noncytolytic, inefficient nAb inducer) and vesicular stomatitis virus (VSV, nonpersisting, cytolytic, potent nAb inducer) transferred the only target of nAb’s from either virus to the other. We analyzed the nAb response to each of the 2 recombinant and parent viruses in infected mice and found that nAb kinetics were solely determined by the viral surface GP and not by the virus backbone. Moreover, the slowly and poorly nAb-triggering LCMV virion was a potent immunogenic matrix for the more antigenic VSV-GP. These findings indicate that the viral GP determines nAb kinetics largely independently of the specific viral infection context. They further suggest that structural features of viral GPs or coevolutionary adaptation of the virus’s GP to the host’s naive B cell repertoire, or both, may critically limit nAb kinetics and improvement of vaccine efficacy.
Authors
Daniel D. Pinschewer, Mar Perez, Eswaraka Jeetendra, Thomas Bächi, Edit Horvath, Hans Hengartner, Michael A. Whitt, Juan Carlos de la Torre, Rolf M. Zinkernagel
×Abstract
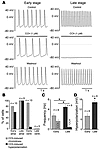
Parasympathetic slowing of the heart rate is predominantly mediated by acetylcholine-dependent activation of the G protein–gated potassium (K+) channel (IK,ACh). This channel is composed of 2 inward-rectifier K+ (Kir) channel subunits, Kir3.1 and Kir3.4, that display distinct functional properties. Here we show that subunit composition of IK,ACh changes during embryonic development. At early stages, IK,ACh is primarily formed by Kir3.1, while in late embryonic and adult cells, Kir3.4 is the predominant subunit. This change in subunit composition results in reduced rectification of IK,ACh, allowing for marked K+ currents over the whole physiological voltage range. As a consequence, IK,ACh is able to generate the membrane hyperpolarization that underlies the strong negative chronotropy occurring in late- but not early-stage atrial cardiomyocytes upon application of muscarinic agonists. Both strong negative chronotropy and membrane hyperpolarization can be induced in early-stage cardiomyocytes by viral overexpression of the mildly rectifying Kir3.4 subunit. Thus, a switch in subunit composition is used to adopt IK,ACh to its functional role in adult cardiomyocytes.
Authors
Bernd K. Fleischmann, Yaqi Duan, Yun Fan, Torsten Schoneberg, Andreas Ehlich, Nibedita Lenka, Serge Viatchenko-Karpinski, Lutz Pott, Juergen Hescheler, Bernd Fakler














Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)