Development

Abstract
Holoprosencephaly (HPE) is a clinically heterogeneous developmental anomaly affecting the CNS and face, in which the embryonic forebrain fails to divide into distinct halves. Numerous genetic loci and environmental factors are implicated in HPE, but mutation in the sonic hedgehog (Shh) gene is an established cause in both humans and mice. As growth arrest–specific 1 (Gas1) encodes a membrane glycoprotein previously identified as a Shh antagonist in the somite, we analyzed the craniofacial phenotype of mice harboring a targeted Gas1 deletion. Gas1–/– mice exhibited microform HPE, including midfacial hypoplasia, premaxillary incisor fusion, and cleft palate, in addition to severe ear defects; however, gross integrity of the forebrain remained intact. These defects were associated with partial loss of Shh signaling in cells at a distance from the source of transcription, suggesting that Gas1 can potentiate hedgehog signaling in the early face. Loss of a single Shh allele in a Gas1–/– background significantly exacerbated the midline craniofacial phenotype, providing genetic evidence that Shh and Gas1 interact. As human GAS1 maps to chromosome 9q21.3–q22, a region previously associated with nonsyndromic cleft palate and congenital deafness, our results establish GAS1 as a potential locus for several human craniofacial malformations.
Authors
Maisa Seppala, Michael J. Depew, David C. Martinelli, Chen-Ming Fan, Paul T. Sharpe, Martyn T. Cobourne

Abstract
Ewing sarcoma gene EWS encodes a putative RNA-binding protein with proposed roles in transcription and splicing, but its physiological role in vivo remains undefined. Here, we have generated Ews-deficient mice and demonstrated that EWS is required for the completion of B cell development and meiosis. Analysis of Ews–/– lymphocytes revealed a cell-autonomous defect in precursor B lymphocyte (pre–B lymphocyte) development. During meiosis, Ews-null spermatocytes were deficient in XY bivalent formation and showed reduced meiotic recombination, resulting in massive apoptosis and complete arrest in gamete maturation. Inactivation of Ews in mouse embryonic fibroblasts resulted in premature cellular senescence, and the mutant animals showed hypersensitivity to ionizing radiation. Finally, we showed that EWS interacts with lamin A/C and that loss of EWS results in a reduced lamin A/C expression. Our findings reveal essential functions for EWS in pre–B cell development and meiosis, with proposed roles in DNA pairing and recombination/repair mechanisms. Furthermore, we demonstrate a novel role of EWS in cellular senescence, possibly through its interaction and modulation of lamin A/C.
Authors
Hongjie Li, Wendy Watford, Cuiling Li, Alissa Parmelee, Mark A. Bryant, Chuxia Deng, John O’Shea, Sean Bong Lee

Abstract
Insig-1 and Insig-2 are regulatory proteins that restrict the cholesterol biosynthetic pathway by preventing proteolytic activation of SREBPs and by enhancing degradation of HMG-CoA reductase. Here, we created Insig–double-knockout (Insig-DKO) mice that are homozygous for null mutations in Insig-1 and Insig-2. After 18.5 days of development, 96% of Insig-DKO embryos had defects in midline facial development, ranging from cleft palate (52%) to complete cleft face (44%). Middle and inner ear structures were abnormal, but teeth and skeletons were normal. The animals were lethargic and runted; they died within 1 day of birth. The livers and heads of Insig-DKO embryos overproduced sterols, causing a marked buildup of sterol intermediates. Treatment of pregnant mice with the HMG-CoA reductase inhibitor lovastatin reduced sterol synthesis in Insig-DKO embryos and reduced the pre-cholesterol intermediates. This treatment ameliorated the clefting syndrome so that 54% of Insig-DKO mice had normal faces, and only 7% had cleft faces. We conclude that buildup of pre-cholesterol sterol intermediates interferes with midline fusion of facial structures in mice. These findings have implications for the pathogenesis of the cleft palate component of Smith-Lemli-Opitz syndrome and other human malformation syndromes in which mutations in enzymes catalyzing steps in cholesterol biosynthesis produce a buildup of sterol intermediates.
Authors
Luke J. Engelking, Bret M. Evers, James A. Richardson, Joseph L. Goldstein, Michael S. Brown, Guosheng Liang

Abstract
Dynamic and reciprocal epithelial-mesenchymal interactions are critical for the normal morphogenesis and maintenance of epithelia. Epimorphin has been identified as a unique molecule expressed by mesenchymal cells and myofibroblasts and has putative morphogenetic effects in multiple epithelial tissues, including intestine, skin, mammary gland, lung, gallbladder, and liver. To define the in vivo role of epimorphin, we created epimorphin-null mice by targeted inactivation of the epimorphin gene. Male epimorphin–/– mice are sterile due to abnormal testicular development and impaired spermatogenesis. Intestinal growth is increased in epimorphin–/– mice due to augmented crypt cell proliferation and crypt fission during the neonatal (suckling) period, mediated at least in part by changes in bone morphogenetic protein (Bmp) and Wnt/β-catenin signaling pathways. Colonic mucosal injury and colitis induced by dextran sodium sulfate (DSS) are ameliorated in epimorphin–/– mice, probably due to the increased proliferative capacity of the epimorphin–/– colon. These in vivo findings support the notion that epimorphin is a key stromal regulator of epithelial cell proliferation and growth in the intestine. In addition, our studies demonstrate a novel and critical role for epimorphin in regulating testicular development and growth as well as spermatogenesis.
Authors
Yuan Wang, Lihua Wang, Hristo Iordanov, Elzbieta A. Swietlicki, Qun Zheng, Shujun Jiang, Yuzhu Tang, Marc S. Levin, Deborah C. Rubin

Abstract
Ectopic pancreas is a developmental anomaly occasionally found in humans. Hes1, a main effector of Notch signaling, regulates the fate and differentiation of many cell types during development. To gain insights into the role of the Notch pathway in pancreatic fate determination, we combined the use of Hes1-knockout mice and lineage tracing employing the Cre/loxP system to specifically mark pancreatic precursor cells and their progeny in Ptf1a-cre and Rosa26 reporter mice. We show that inactivation of Hes1 induces misexpression of Ptf1a in discrete regions of the primitive stomach and duodenum and throughout the common bile duct. All ectopic Ptf1a-expressing cells were reprogrammed, or transcommitted, to multipotent pancreatic progenitor status and subsequently differentiated into mature pancreatic exocrine, endocrine, and duct cells. This process recapitulated normal pancreatogenesis in terms of morphological and genetic features. Furthermore, analysis of Hes1/Ptf1a double mutants revealed that ectopic Ptf1a-cre lineage–labeled cells adopted the fate of region-appropriate gut epithelium or endocrine cells similarly to Ptf1a-inactivated cells in the native pancreatic buds. Our data demonstrate that the Hes1-mediated Notch pathway is required for region-appropriate specification of pancreas in the developing foregut endoderm through regulation of Ptf1a expression, providing novel insight into the pathogenesis of ectopic pancreas development in a mouse model.
Authors
Akihisa Fukuda, Yoshiya Kawaguchi, Kenichiro Furuyama, Sota Kodama, Masashi Horiguchi, Takeshi Kuhara, Masayuki Koizumi, Daniel F. Boyer, Koji Fujimoto, Ryuichiro Doi, Ryoichiro Kageyama, Christopher V.E. Wright, Tsutomu Chiba

Abstract
Ikaros transcription factors are essential regulators of lymphopoiesis and the development of the immune system. We now show that Ikaros is expressed in hormone-producing pituitary corticomelanotroph cells, where it binds the proopiomelanocortin promoter and regulates endogenous gene expression. Loss of Ikaros in vivo results in contraction of the pituitary corticomelanotroph population, reduced circulating adrenocorticotrophic hormone levels, and adrenal glucocorticoid insufficiency. While hemopoietic reconstitution failed to correct this hormonal deficit, the phenotype of reduced body weight and diminished survival was rescued by systemic glucocorticoid-hormone administration. Given the established immunomodulatory properties of glucocorticoid hormones, these findings reveal a novel role for Ikaros in orchestrating immune-endocrine development and function.
Authors
Shereen Ezzat, Rene Mader, ShunJiang Yu, Terry Ning, Philippe Poussier, Sylvia L. Asa

Abstract
B lymphocyte differentiation is coordinated with the induction of high-level Ig secretion and expansion of the secretory pathway. Upon accumulation of unfolded proteins in the lumen of the ER, cells activate an intracellular signaling pathway termed the unfolded protein response (UPR). Two major proximal sensors of the UPR are inositol-requiring enzyme 1α (IRE1α), an ER transmembrane protein kinase/endoribonuclease, and ER-resident eukaryotic translation initiation factor 2α (eIF2α) kinase (PERK). To elucidate whether the UPR plays an important role in lymphopoiesis, we carried out reconstitution of recombinase-activating gene 2–deficient (rag2–/–) mice with hematopoietic cells defective in either IRE1α- or PERK-mediated signaling. IRE1α-deficient (ire1α–/–) HSCs can proliferate and give rise to pro–B cells that home to bone marrow. However, IRE1α, but not its catalytic activities, is required for Ig gene rearrangement and production of B cell receptors (BCRs). Analysis of rag2–/– mice transplanted with IRE1α trans-dominant-negative bone marrow cells demonstrated an additional requirement for IRE1α in B lymphopoiesis: both the IRE1α kinase and RNase catalytic activities are required to splice the mRNA encoding X-box–binding protein 1 (XBP1) for terminal differentiation of mature B cells into antibody-secreting plasma cells. Furthermore, UPR-mediated translational control through eIF2α phosphorylation is not required for B lymphocyte maturation and/or plasma cell differentiation. These results suggest specific requirements of the IRE1α-mediated UPR subpathway in the early and late stages of B lymphopoiesis.
Authors
Kezhong Zhang, Hetty N. Wong, Benbo Song, Corey N. Miller, Donalyn Scheuner, Randal J. Kaufman
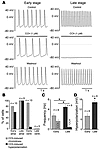
Abstract
Parasympathetic slowing of the heart rate is predominantly mediated by acetylcholine-dependent activation of the G protein–gated potassium (K+) channel (IK,ACh). This channel is composed of 2 inward-rectifier K+ (Kir) channel subunits, Kir3.1 and Kir3.4, that display distinct functional properties. Here we show that subunit composition of IK,ACh changes during embryonic development. At early stages, IK,ACh is primarily formed by Kir3.1, while in late embryonic and adult cells, Kir3.4 is the predominant subunit. This change in subunit composition results in reduced rectification of IK,ACh, allowing for marked K+ currents over the whole physiological voltage range. As a consequence, IK,ACh is able to generate the membrane hyperpolarization that underlies the strong negative chronotropy occurring in late- but not early-stage atrial cardiomyocytes upon application of muscarinic agonists. Both strong negative chronotropy and membrane hyperpolarization can be induced in early-stage cardiomyocytes by viral overexpression of the mildly rectifying Kir3.4 subunit. Thus, a switch in subunit composition is used to adopt IK,ACh to its functional role in adult cardiomyocytes.
Authors
Bernd K. Fleischmann, Yaqi Duan, Yun Fan, Torsten Schoneberg, Andreas Ehlich, Nibedita Lenka, Serge Viatchenko-Karpinski, Lutz Pott, Juergen Hescheler, Bernd Fakler

Abstract
One of the most perplexing questions in clinical genetics is why patients with identical gene mutations oftentimes exhibit radically different clinical features. This inconsistency between genotype and phenotype is illustrated in the malformation spectrum of holoprosencephaly (HPE). Family members carrying identical mutations in sonic hedgehog (SHH) can exhibit a variety of facial features ranging from cyclopia to subtle midline asymmetries. Such intrafamilial variability may arise from environmental factors acting in conjunction with gene mutations that collectively reduce SHH activity below a critical threshold. We undertook a series of experiments to test the hypothesis that modifying the activity of the SHH signaling pathway at discrete periods of embryonic development could account for the phenotypic spectrum of HPE. Exposing avian embryos to cyclopamine during critical periods of craniofacial development recreated a continuum of HPE-related defects. The craniofacial malformations included hypotelorism, midfacial hypoplasia, and facial clefting and were not the result of excessive crest cell apoptosis. Rather, they resulted from molecular reprogramming of an organizing center whose activity controls outgrowth and patterning of the mid and upper face. Collectively, these data reveal one mechanism by which the variable expressivity of a disorder such as HPE can be produced through temporal disruption of a single molecular pathway.
Authors
Dwight Cordero, Ralph Marcucio, Diane Hu, William Gaffield, Minal Tapadia, Jill A. Helms

Abstract
Classical research has suggested that early palate formation develops via epithelial-mesenchymal interactions, and in this study we reveal which signals control this process. Using Fgf10–/–, FGF receptor 2b–/– (Fgfr2b–/–), and Sonic hedgehog (Shh) mutant mice, which all exhibit cleft palate, we show that Shh is a downstream target of Fgf10/Fgfr2b signaling. Our results demonstrate that mesenchymal Fgf10 regulates the epithelial expression of Shh, which in turn signals back to the mesenchyme. This was confirmed by demonstrating that cell proliferation is decreased not only in the palatal epithelium but also in the mesenchyme of Fgfr2b–/– mice. These results reveal a new role for Fgf signaling in mammalian palate development. We show that coordinated epithelial-mesenchymal interactions are essential during the initial stages of palate development and require an Fgf-Shh signaling network.
Authors
Ritva Rice, Bradley Spencer-Dene, Elaine C. Connor, Amel Gritli-Linde, Andrew P. McMahon, Clive Dickson, Irma Thesleff, David P.C. Rice
No posts were found with this tag.



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)