Comments for:

Abstract
Missense mutations in perforin, a critical effector of lymphocyte cytotoxicity, lead to a spectrum of diseases, from familial hemophagocytic lymphohistiocytosis to an increased risk of tumorigenesis. Understanding of the impact of mutations has been limited by an inability to express human perforin in vitro. We have shown, for the first time to our knowledge, that recombinant human perforin is expressed, processed appropriately, and functional in rat basophilic leukemia (RBL) cells following retroviral transduction. Subsequently, we have addressed how perforin missense mutations lead to absent perforin detection and impaired cytotoxicity by analyzing 21 missense mutations by flow cytometry, immunohistochemistry, and immunoblot. We identified perforin missense mutations with partial maturation (class 1), no apparent proteolytic maturation (class 2), and no recognizable forms of perforin (class 3). Class 1 mutations exhibit lytic function when expressed in RBL cells and are associated with residual protein detection and variable cytotoxic function in affected individuals, suggesting that carriers of class 1 alleles may exhibit more subtle immune defects. By contrast, class 3 mutations cause severely diminished perforin detection and cytotoxicity, while class 2 mutations have an intermediate phenotype. Thus, the pathologic mechanism of perforin missense mutation likely involves a protein dosage effect of the mature protein.
Authors
Kimberly A. Risma, Robert W. Frayer, Alexandra H. Filipovich, Janos Sumegi
Response from the authors
Submitter: Janos Sumegi | Janos.Sumegi@cchmc.org
Cincinnati Children's Hospital Medical Center, University of Cincinnati, Faculty of Medicine
Published February 15, 2006
We wish to thank Drs. Trapani and Voskoboinik for their interest in our manuscript “Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis” (1). We appreciate their detailed, critical review and would like to address their concerns.
1. We have published, for the first time to our knowledge, data directly comparing the expression and processing of human and mouse perforin expressed in rat basophilic leukemia (RBL) cells (1). Using equal amounts of total cellular protein, the native mouse and human perforin were equally detectable by immunoblot (non-reducing gel) and showed CMA-sensitive proteolytic maturation in RBL-2H3 cells (Figure 2). Upon comparison of immunoblots of human, rat, and mouse perforin expressed in RBL cells, we concluded that the human and rat displayed similar banding patterns, but the mouse perforin showed delayed mobility. There is no evidence for abnormal processing of human perforin in RBL cells as suggested by Drs. Trapani and Voskoboinik. In fact, we have shown that the processing of human perforin in RBL-1, RBL-2H3, and NK92 cells is indistinguishable by the same methods described by Uellner et al. (2): (a) processing is inhibited by concanamycin A and leupeptin; (b) the mature form is present in dense granules; and (c) the mature form of perforin is lytic. In Figure 3 we have shown data comparing the functional activity of perforin from both species. Although mouse perforin shows higher lytic activity than human perforin when measuring rbc lysis, the differences are not 5-fold as described by Drs. Trapani and Voskoboinik. Over 4 independent lytic assays, we have measured chromium release from rbc’s as a measure of perforin function. In our studies, human perforin showed consistently 60%–70% of the activity of mouse perforin (see Supplemental Figure 2, provided below). We have also sorted RBL-2H3 cells by GFP expression prior to functional studies; however, we did not normalize our data to the number of GFP expressing cells, because the levels of native perforin were equivalent when comparing equal amounts of protein by immunoblot as described above (Figure 2). The lytic activity of human perforin was reduced in Figure 5B compared to 3A as noted. These experiments were independent retroviral transductions and the efficiency of transduction was reduced in the second experiment. Mouse perforin was not included in the second retroviral transduction, preventing direct comparison. Our results differed significantly from those of Uellner et al., who showed that very little mature human perforin was detectable following stable transfection of RBL cells (2). The methodology for achieving cDNA expression was different – they generated stable transfectants after electroporation and selection in antibiotic-containing media. This method was unsuccessful in our hands as well. We found, as did Drs. Trapani and Voskoboinik (3-5), that the use of retroviral transduction of RBL cells was superior.
2. The RBL-1 cell line is derived from the same rat leukemia cells as RBL-2H3 and was selected by the inability to release histamine upon IgE coupled degranulation (6). This defective secretory capacity was confirmed in Gao et al.’s studies (7) and ours as well. However, Gao et al. also showed that proteolytic processing of a recombinant protein targeted for secretory lysosomes was equivalent between the two cell lines as we have also shown for recombinant perforin (Figures 1 and 5). To address the specific concern for detection of PRF1-R225W and PRF1-G149S, we would like to clarify that the full-length protein is present and detectable by both H315 and P1-8 antibodies under non-reducing conditions in Figure 8A. The full-length PRF1-R225W and PRF1-G149S in Figure 8 illustrate more extensive degradation than in the Supplemental Figure (as detected under reducing conditions). This reiterates why we favor the use of non-reducing conditions to analyze perforin by Western blot. Under these conditions a native conformation of the protein is detected, and misfolded proteins are retained in high molecular weight aggregates. It is true that by Western blot, under reducing conditions, many of the mutants were detected more readily than wild-type perforin. However, this is not seen under native conditions used for flow cytometry, immunohistochemistry, and non-reducing SDS-PAGE. We believe the latter methods detect properly folded perforin; whereas, immunoblots performed under reducing conditions allow detection of both misfolded and native protein.
3. We agree that more extensive functional studies are required to test our hypothesis that perforin function is dependent upon the presence of the mature form. We were pleased that our results from testing the function of mutant human perforins expressed in RBL-2H3 cells were consistent with the findings of Drs. Trapani and Voskoboinik (3-5). We agree that there will be post-synaptic functional changes, as they have described for G429E (3).
4. We are aware that there are conserved domains with high homology between human and mouse perforin; however, in the absence of a 3- dimensional structure for perforin it is not possible to predict with confidence which residues result in conformational changes between human and mouse perforin.
To summarize, there are three main differences between our paper and the studies of Drs. Voskoboinik and Trapani (1, 3-5). First, we have shown that the expression of functional, mature human perforin is readily studied in the RBL cell line and conclude that this is an appropriate model to evaluate the impact of perforin mutations related to hemophagocytic lympho- histiocytosis. Second, we utilized a novel approach to study the biochemical structure of human perforin by achieving high-level expression of human perforin in RBL-1 cells. Third, we focused on the maturation of human perforin with and without mutations and found that classification of mutations according to proteolytic processing and folding was correlative with the age of onset of hemophagocytic lymphohistiocytosis. In contrast, the important work provided by Drs. Voskoboinik and Trapani has provided extensive functional analysis of recombinant mouse perforin with and without mutations. Comparison of these complementary studies has greatly increased our understanding of perforin biology. We agree that the most appropriate methodology to study perforin is in the context of the cytotoxic lymphocyte and these experiments will yield important insight into the “vexed question of assessing perforin expression, processing and function”.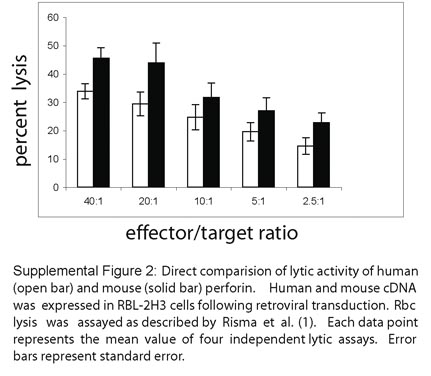
Kimberly A. Risma, M.D., Ph.D, and Janos Sumegi, M.D., Ph.D
Department of Allergy/Immunology
Department of Hematology/Oncology
Cincinnati Children’s Hospital Medical Center
University of Cincinnati College of Medicine
Cincinnati, Ohio, USA
1. Risma, K.A., Frayer, R.W., Filipovich, A.H., and Sumegi, J. 2006. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J. Clin. Invest. 116:182-192.
2. Uellner, R., Zvelebil, M.J., Hopkins, J., Jones, J., Mac-Dougall, L.K., Morgan, B.P., Podack, E., Waterfield, M.D., and Griffiths, G.M. 1997. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J. 16:7287-7296.
3. Voskoboinik, I., Thia, M.C., De Bono, A., Browne, K., Cretney, E., Jackson, J.T., Darcy, P.K., Jane, S.M., Smyth, M.J., and Trapani, J.A.. 2004. The functional basis for Hemophagocytic Lymphohistiocytosis in a patient with co-inherited missense mutations in the perforin (PFN1) gene. J. Exp. Med. 200:811-816.
4. Voskoboinik, I., Thia, M.C., Fletcher, J., Ciccone, A., Browne, K., Smyth, M.J., and Trapani, J.A.. 2005. Calcium-dependent plasma membrane binding and cell lysis by perforin are mediated through its C2 domain. A critical role for aspartate residues 429, 435, 483, and 485 but not 491. J Biol. Chem 280: 8426-8434.
5. Voskoboinik, I., Thia, M.C., and Trapani, J.A. 2005. A functional analysis of the putative polymorphisms A91V and N252S, and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood. 105: 4700-4706
6. Barsumian, E.L., Isersky, C., Petrino, M.G, and Siraganian, R.P. 1981. IgE- induced histamine release from rat basophilic leukemia cell lines: isolation of releasing and nonreleasing clones. Eur. J. Immunol. 11:317-323.
7. Gao, Y., Hansson, M., Calafat, J., Tapper, H., Olsson, I.. 2004. Sorting soluble tumor necrosis factor (TNF) receptor for storage and regulated secretion in hematopoietic cells. J Leukoc Biol. 76:876-85.
Janos Sumegi, M.D., Ph.D
Professor of Pediatrics
Division of Hematology and Oncology
Cincinnati Children's Hospital Medical Center
University of Cincinnati, Faculty of Medicine
3333 Burnett Ave.
Cincinnati, Ohio 45229
phone: 513-636-5976
fax: 513-636-3549
The vexed question of assessing perforin expression, processing and function
Submitter: Joseph A. Trapani | joe.trapani@petermac.org
Peter MacCallum Cancer Centre
Published February 15, 2006
The vexed question of assessing perforin expression, processing and function
Joseph A. Trapani and Ilia Voskoboinik
Cancer Immunology Program, Peter MacCallum Cancer Centre, St. Andrew’s Place, East Melbourne, 3002, Australia.
Ph.: (61 3) 9656 3726 Fax.: (61 3) 9656 1411 E-mail: joe.trapani@petermac.org
Congenital deficiency of the lymphocyte pore-forming protein perforin, which is often caused by missense mutations of both perforin alleles, results in fatal familial haemophagocytic lymphohistiocytosis (FHL) in affected infants. We have recently investigated the functional basis for most of the missense mutations identified in FHL patients using recombinant expression of perforin in rat basophil leukemia (RBL-2H3) cellssupported by independent experiments using baculovirus-expressed and purified perforin. We explored both the molecular basis of FHL-related immunodeficiency and assigned molecular function (such as Ca2+-dependent target cell membrane binding) to specific perforin residues and its putative functional domains (1-3).
A study that also used RBL expression was recently reported in JCI by Risma et al. (4), the main difference being that human perforin was expressed, whereas we elected to “graft” missense mutations onto the mouse perforin sequence. Risma et al. questioned our use of mouse (rather than human) protein, and strongly implied that some of our conclusions are unsafe. The authors also proposed a new classification of perforin mutations, based solely on the post-translational processing of the mutated proteins. We strongly disagree with Risma et al's conclusions on our previous findings, and in turn have problems with their methodologies and conclusions. We offer the following comments:
1.As a prelude to our reported studies, we carefully compared the expression and function of human and mouse perforin in RBL cells and found that the lytic efficiency of mouse perforin (calculated by comparing the number of RBL cells needed to achieve a given level of target cell lysis) is ~5- fold higher than human. As purified mouse and human perforin have similar intrinsic activity, it can be concluded that mouse perforin is more efficiently expressed, processed and/or trafficked in rodent RBL cells. Risma et al indeed report similar findings (eg, compare the maximum levels of cytotoxicity in Fig. 3A and 5B) which they do not discuss. Furthermore, the original paper that compared perforin processing in human YT cells and RBL ((5) also cited by Risma et al.) using a range of monoclonal antibodies, clearly indicated that human perforin is poorly processed in RBL cells as opposed to human NK cells. We therefore advise caution in interpreting data on human perforin processing in rodent RBL cells.
2.Risma et al. used the RBL variant RBL-1 (which is incapable of target cell killing due to a lack of perforin activation or secretion) together with various antibodies used in immunoblotting to study perforin processing rather than RBL-2H3 cells, which were used in cytotoxicity assays. But Gao et al ((6) also cited by Risma et al.) described dissimilar compartmentalisation of proteins by the two cell lines, suggesting that perforin is processed differently. To exclude this possibility, appropriate antibodies such as those validated by Uellner et al (5) should have been used. Rather, Risma et al have extensively used the anti-mouse perforin antibody P1-8 or the anti-human H315 antibody, leading to considerable inconsistency and confusion. For example, in Figure 8A R225W appears to be truncated when probed with H315, but some full-length protein is seen when P1-8 is used. The authors also failed to acknowledge that their finding of truncated R225W perforin agreed with ours (1). Similarly, G149S shows no full-length protein in Fig 8A, but it is clearly visible on the Supplementary Figure.
3.Risma et al examined the immunoblotting pattern of many mutants, and on this basis proposed that all perforin mutations result in various degrees of degradation and inadequate processing. However, they supported this hypothesis with functional (lysis) data on only four variants (Fig. 5B), two of which (N252S and A91V) are polymorphisms. In each case, their results agreed with ours: wild-type function for N252S, 50% function for A91V and total loss of function for R225W and C73R. It puzzles us that the authors can draw a general conclusion on the functional effects of perforin mutation from so few examples. By contrast, we have assessed the function of more than twenty mutants (3) and more than a dozen additional missense mutations affecting the C2 domain (2). We disagree that it is not necessary to sort RBL cells with equivalent levels of GFP fluorescence prior to assessing perforin expression and processing (7), perhaps accounting for the fact that the great majority of the mutations studied by Risma et al appear to be expressed at much higher levels than wild-type perforin in RBL-1 cells (Supplementary Figure), a curious finding. In fact, as pointed out by the authors themselves, FHL patients almost invariably show markedly reduced perforin expression in their cytolytic lymphocytes (compare Table I, Figure 4 and Supplementary Figure).
Our work was criticized because we claimed no absolute correlation between perforin expression level and cytotoxic function. But Risma et al. have ignored the fact that some FHL-associated mutations (1, 3) and many experimental mutations (eg. in the calcium binding domain, (2)) result in normal expression, storage and secretion of perforin, but total loss of function. Thus Risma et al. neglect the possibility that perforin mutations may affect not only its pre-synaptic processing and trafficking, but also its post- synaptic function, for example as has been clearly shown by us for the G428E mutation (1).
4.Risma et al. also argue that mouse perforin may not be suitable for studying human perforin mutations because around 30% of their amino acid residues are not identical. But the authors ignore the fact that the non- identical residues are frequently conservative substitutions and that the putative domain boundaries are tightly conserved. Our experiments were carefully controlled in that all of the known variations in amino acid sequence at a given position between rodents, human and the very distant species such as the flounder were tested in our functional assays (3). Without exception, substitution of the corresponding wild-type human or flounder residue into mouse perforin resulted in completely normal processing and cytotoxicity.
In conclusion, we are fully aware of the limitations of the RBL system, but we totally disagree that only human perforin can be informative in that setting. Ultimately, methodologies for re-introducing perforin expression into perforin-null primay lymphocytes are sorely needed, and several groups are currently pursuing this goal.
References
1.Voskoboinik, I., Thia, M.-C., De Bono, A., Browne, K., Cretney, E., Jackson, J.T., Darcy, P.K., Jane, S.M., Smyth, M.J., and Trapani, J.A. 2004. The functional basis for hemophagocytic lymphohistiocytosis in a patient with co- inherited missense mutations in the perforin (PFN1) gene. J Exp Med 200:811-816.
2.Voskoboinik, I., Thia, M.C., Fletcher, J., Ciccone, A., Browne, K., Smyth, M.J., and Trapani, J.A. 2005. Calcium-dependent plasma membrane binding and cell lysis by perforin are mediated through its C2 domain: A critical role for aspartate residues 429, 435, 483, and 485 but not 491. J Biol Chem 280:8426-8434.
3.Voskoboinik, I., Thia, M.C., and Trapani, J.A. 2005. A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood 105:4700-4706.
4.Risma, K.A., Frayer, R.W., Filipovich, A.H., and Sumegi, J. 2006. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J Clin Invest 116:182-192.
5.Uellner, R., Zvelebil, M.J., Hopkins, J., Jones, J., MacDougall, L.K., Morgan, B.P., Podack, E., Waterfield, M.D., and Griffiths, G.M. 1997. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. Embo J 16:7287-7296.
6.Gao, Y., Hansson, M., Calafat, J., Tapper, H., and Olsson, I. 2004. Sorting soluble tumor necrosis factor (TNF) receptor for storage and regulated secretion in hematopoietic cells. J Leukoc Biol 76:876-885.
7.Voskoboinik, I., and Trapani, J.A. 2006. Addressing the mysteries of perforin function. Immunol Cell Biol 84:66-71.



Copyright © 2024 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)