Issue published March 2, 2026 Previous issue

- Volume 136, Issue 5
Go to section:
On the cover: Dual role for macrophages in muscle regeneration and degeneration
Yin et al. report that a macrophage subset expressing activin A facilitates muscle regeneration by acting on satellite cells but contributes to heterotopic ossification in pathological settings such as fibrodysplasia ossificans progressiva. The cover image shows injured skeletal muscle sections immunostained with anti–mouse embryonic myosin heavy chain (eMyHC) (magenta), marking regenerating muscle fibers; mouse IgG (light blue), indicating necrotic fibers; and Hoechst (yellow), labeling nuclei. Image credit: Wenqiang Yin, Kazuo Okamoto, and Hiroshi Takayanagi.

-
Reviews
×
Abstract
Diabetic retinopathy (DR), the most common microvascular complication in patients with diabetes mellitus (DM), is a leading cause of vision loss worldwide. Sustained hyperglycemia plays a central role in promoting DR. However, tight glycemic control does not prevent — and indeed sometimes worsens — DR, highlighting the importance of ongoing studies aimed at improving our understanding of this complex disease. Over the last few decades, the dogma that DR is a vascular disease that results in secondary neuronal injury has evolved, as emerging evidence suggests that neurodegeneration occurs in parallel with or prior to vascular cell injury in the retina of patients with DM. This has led to appreciation of DR as a neurovascular disease, characterized by microvascular injury and neurodegeneration, both of which contribute to vision loss. Here, we explore how molecular stress (i.e., glucose dysregulation, dysmetabolism, oxidative stress, and inflammation) promote retinal vascular cell and neuronal injury in patients with DM. We focus on how these processes influence, and are influenced by, genes regulated by the HIF family of transcription factors in glial, vascular, neuronal, and inflammatory cells, with the goal of identifying new therapeutic avenues for the prevention or early treatment of patients with this vision-threating disease.
Authors
Chuanyu Guo, Akrit Sodhi
×Abstract
Urothelial cancers of the urinary tract are the fourth most common malignancy in men, with a shifting demographic affecting younger patients and an increasing incidence in females. In this Review, we discuss recent discoveries and paradigm-shifting clinical trials that impact all stages of urothelial cancer. New therapeutics and drug-delivery devices have led to multiple approvals for treatments of non-muscle invasive bladder cancer. The addition of chemotherapy, immunotherapy, and antibody-drug conjugates is transforming perioperative treatment for patients with muscle-invasive bladder cancer. The use of liquid biomarkers, circulating tumor DNA, and urinary tumor DNA is aiding the identification of patients at risk for local recurrence and possibly those who can avoid systemic therapy. Finally, integrating biomarkers and systemic treatments is creating a paradigm that could lead to the successful treatment of bladder cancer without requiring bladder removal. Overall, these advancements in biomarkers and novel therapeutics are likely to dramatically improve survival for bladder cancer.
Authors
Joshua J. Meeks


-
Commentaries
×
Abstract
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive sarcomas that constitute a major cause of mortality in individuals with neurofibromatosis type 1 (NF-1) and exhibit highly variable responses to radiotherapy. In this issue of the JCI, Zhu and colleagues integrated functional genomics, single-cell transcriptomics, and analysis of human tumors to show that type I IFN signaling shapes both tumor-intrinsic radiation sensitivity of MPNSTs and local recruitment and activation of T cells. Their findings establish IFN signaling as a central coordinator of the radiotherapy response in MPNSTs and suggest that incorporating targeted immunomodulation strategies may improve radiotherapy outcomes. The work also has direct implications for the role of the immune system and IFN signaling radiation–based treatment of soft tissue sarcomas beyond those involved in NF-1.
Authors
Sean P. Pitroda, Ralph R. Weichselbaum
×Abstract
Organized adaptive immunity can emerge in the CNS under specific inflammatory and stromal conditions. The study by Yang et al. in this issue of the JCI reports that experimental ischemic stroke induced germinal center–like B cell follicles through microglial MIF–CD74/CXCR4 signaling and in situ B cell proliferation, promoting chronic neuroinflammation. These findings align with a growing body of evidence that the brain and meninges can support ectopic lymphoid structures in multiple sclerosis, during aging, and in certain gliomas. This Commentary integrates these observations to highlight shared principles, disease-specific outcomes, and unresolved questions regarding the identity and function of lymphoid aggregates in the CNS.
Authors
Catalina Lee-Chang
×Abstract
Immunotherapy has shown limited efficacy in glioblastoma (GBM), reflecting profound immune evasion and an immunosuppressive microenvironment. In this Commentary, we highlight recent work by Zhang and colleagues identifying the transcription factor OLIG2 as a central mediator of immune evasion in GBM. Though OLIG2 has an established role in promoting GBM progression through its effects on glioma stem-like cells (GSCs), Zhang et al. demonstrated a further role for OLIG2 in suppressing antitumor immunity: in human GSCs and GSCs from mouse models of GBM, OLIG2 expression epigenetically repressed the interferon-responsive chemokine CXCL10, thereby limiting cytotoxic T cell infiltration. These findings provide a mechanistic explanation for immune resistance in GBM and support targeting tumor-intrinsic chromatin programs to enhance responses to immunotherapy.
Authors
Raymond Sun, Chao Gao, Rongze Olivia Lu
×Safeguarding lymphatic identity: cooperative Erg and Fli1 activity in lymphatic vascular homeostasis
Abstract
Although transcriptional programs driving lymphatic endothelial cell (LEC) specification are being increasingly characterized, far less is known about the postnatal mechanisms that preserve lymphatic vessel identity and function. In this issue of the JCI, Yang et al. show that the E26 transformation-specific (ETS) transcription factors ETS-related gene (Erg) and Friend leukemia integration 1 (Fli1) cooperatively maintain adult LEC homeostasis by sustaining transcriptionally distinct LEC populations, vascular integrity, immune-vascular interactions, and repression of proinflammatory and prothrombotic gene programs. These findings extend the known roles of Erg and Fli1 beyond the blood endothelium and provide mechanistic insight into human lymphatic disease associated with Erg haploinsufficiency.
Authors
Kelly de Korodi, Tatiana V. Petrova

-
Research Letter
×
Abstract
Authors
Robert Corty, Yash Pershad, J. Brett Heimlich, Caitlyn Vlasschaert, Leo Luo, Taralynn Mack, Kaushik Amancherla, Cassianne Robinson-Cohen, Michael Savona, Alexander G. Bick
-
Research Articles
×
Abstract
The link between glutaminolysis and osteoarthritis (OA) has only recently begun to be elucidated. Here, we report the association of obesity- and injury-induced cartilage damage with impaired glutaminolysis in chondrocytes. Defective glutaminolysis triggered the onset and progression of OA, with enhanced catabolism and decreased anabolism. Supplementation of α-ketoglutarate (αKG), a key component in glutaminolysis and an epigenetic factor, effectively protected cartilage against degradation in vivo via a TCA cycle– and HIF-1α–independent manner. Mechanistically, OA pathogenic factors increased H3K27me3 deposition on promoters of key glutaminolysis genes, including Slc1a5 and Gls1, leading to impaired glutaminolysis. Conversely, αKG facilitated Kdm6b-dependent H3K27me3 demethylation of not only glutaminolysis genes to rescue Gln metabolism but also Ube2o to reverse OA. Elevated Ube2o expression led to TRAF6 ubiquitination and subsequent inhibition of NF-κB signaling, thereby reversing the pathological reprogramming of glycolysis and oxidative phosphorylation and protecting against cartilage destruction. Collectively, these results demonstrated that OA pathogenic factors impair glutaminolysis through epigenetic regulation, which further exacerbate OA. Moreover, αKG restores metabolic homeostasis and alleviates OA through H3K27me3 demethylation.
Authors
Shuaijun Li, Jiefeng Huang, Ting Shang, Laiya Lu, Orion R. Fan, Peisheng Jin, Xin Zou, Zixin Cai, Wuyan Lu, Shuangmeng Jia, Linxiao Li, Ke Fang, Fengting Niu, Jiaojiao Li, Cheng Zhao, Qian Wang, Ruizhu Sun, Si Shi, Feng Yin, Yun Zhang, Yi Eve Sun, Lei Cui
×Abstract
MRE11, a breast tumor suppressor and component of the MRE11-RAD50-NBS1 complex, plays a critical role in DNA end resection and initiation of ataxia-telangiectasia mutation–dependent (ATM-dependent) DNA damage signaling. However, the precise mechanisms governing MRE11 function in the DNA damage response (DDR) remain incompletely understood. Here, we found that MRE11 is deacetylated by the SIRT2 sirtuin deacetylase and breast tumor suppressor, which promotes DNA binding to facilitate DNA end resection and ATM-dependent signaling. SIRT2 deacetylase activity promoted DNA end resection. SIRT2 further complexed with and deacetylated MRE11 at conserved lysine 393 (K393) in response to DNA double-stranded breaks (DSBs), which promoted MRE11 localization and DNA binding at DSBs but not interaction with RAD50, NBS1, or CtIP. Moreover, MRE11 K393 deacetylation by SIRT2 promoted ATM-dependent signaling. Our findings define a mechanism regulating MRE11 binding to DNA through SIRT2 deacetylation, elucidating a critical upstream signaling event directing MRE11 function in the DDR and providing insight into how SIRT2 dysregulation leads to genomic instability and tumorigenesis.
Authors
Fatmata Sesay, Hui Zhang, Priya Kapoor-Vazirani, Andrew T. Jung, Mark E. Essien, Amanda J. Bastien, Nho C. Luong, Xu Liu, PamelaSara E. Head, Duc M. Duong, Xiaofeng Yang, Zachary S. Buchwald, Xingming Deng, Nicholas T. Seyfried, David S. Yu
×Abstract
The multi-omics data represented by genomic data from patients with metastatic triple-negative breast cancer (TNBC) is crucial for precision treatment, yet data on genomic alterations in metastatic cohorts and Chinese populations remains limited. We performed targeted sequencing of 296 metastatic TNBC samples from 296 patients treated at Fudan University Shanghai Cancer Center (October 2018 to November 2020) using a 484-gene panel, identifying 796 metastatic events across 18 organ sites. We characterized the genomic landscape of TNBC metastases and identified marked enrichment of polycystin-1 (PKD1) mutations in metastatic lesions — a finding validated in an independent paired primary metastasis cohort (n = 105). Notably, PKD1 mutations were associated with resistance to anti–PD-1 therapy, as validated across 3 clinical trials (NCT03805399, NCT04129996, and NCT04395989). Multi-omics analyses, combined with functional in vitro and in vivo mechanistic studies, revealed that PKD1 modulated the “desert” tumor immune microenvironment via C-C motif chemokine ligand 2 (CCL2), and targeting CCL2 could reverse immunotherapy resistance. This comprehensive genomic characterization of metastases enhances our understanding of tumor evolution, identifies PKD1 as a previously uncharacterized regulator of immune evasion to our knowledge, and suggests a potential therapeutic strategy to overcome immunotherapy resistance.
Authors
Xiu-Zhi Zhu, Yi-Fan Zhou, Xiao-Han Ying, Yun-Yi Wang, Xiao-Hong Ding, Kun-Yu Zhang, Zhi-Ming Shao, Xi Jin, Yi-Zhou Jiang, Zhong-Hua Wang
×Abstract
Large-cohort GWAS for alcohol use disorder (AUD) drug treatment outcomes and AUD risk have repeatedly identified genetic loci that are splicing quantitative trait loci for the fibronectin III domain containing 4 (FNDC4) gene in the brain. However, FNDC4 function in the brain and how it might contribute to AUD pathophysiology remain unclear. In the present study, we characterized GWAS loci–associated FNDC4 splice isoforms and demonstrated that FNDC4 alternative splicing results in loss of function for FNDC4. We also investigated FNDC4 function using CRISPR/Cas9 editing and the creation of human induced pluripotent stem cell–derived (iPSC-derived) neural organoids joined with single-nucleus RNA sequencing, a series of studies that showed that FNDC4 KO resulted in a striking shift in the relative proportions of glutamatergic and GABAergic neurons in iPSC-derived forebrain organoids as well as changes in their electrical activity. We further explored a potential mechanism(s) of FNDC4-dependent neurogenesis, and the results suggested a role for FNDC4 in mediating neural cell surface interactions. In summary, this series of experiments indicates that FNDC4 plays a role in regulating cerebral cortical neurogenesis in the brain. This regulation may contribute to the response to AUD pharmacotherapy as well as the effects of alcohol on the brain.
Authors
Xiujuan Zhu, August J. John, Sooan Kim, Li Wang, Enci Ding, Jing Zheng, Ateka Saleh, Irene Marín-Goñi, Abedalrahman Jomaa, Huanyao Gao, Meijie Wang, Ching Man Wai, Irene Moon, Cindy Chen, Alireza Agahi, Brandon J. Coombes, Tony M. Kerr, Nobuyoshi Suto, Liewei Wang, Mark A. Frye, Joanna M. Biernacka, Victor M. Karpyak, Hu Li, Richard M. Weinshilboum, Duan Liu
×Abstract
The lymphatic system maintains tissue fluid balance, and FOXC2 mutations cause lymphoedema-distichiasis syndrome, which is characterized by lymphatic valve defects. Although oscillatory shear stress regulates FOXC2 expression, other extracellular regulators remain unclear. In this study, we identified LPA4 and LPA6, two Gα12/Gα13-coupled receptors for the bioactive lipid lysophosphatidic acid (LPA), as key regulators of FOXC2 expression and lymphatic valve development. Lymphatic endothelial cell–specific (LEC-specific) Lpa4 Lpa6–deficient mice exhibited impaired lymphatic valve formation and maintenance, which resembled phenotypes of LEC-specific Foxc2-deficient mice, including abnormal lymphatic vessel patterning. Mechanistically, lymphatic endothelial Lpa4/Lpa6 ablation reduced FOXC2 expression in vitro and in vivo. NF-κB was found to be essential for LPA-induced FOXC2 expression through the LPA4/LPA6-Gα12/Gα13-Rho kinase signaling axis. Accordingly, pharmacological inhibition of NF-κB and Rho kinase impaired lymphatic valve maintenance in mice. These results suggested that lymphatic endothelial LPA4 and LPA6 synergistically regulate FOXC2 expression through NF-κB activation and play an important role in lymphatic valve formation and maintenance. Our findings provide a molecular basis for lymphatic vessel development with a therapeutic potential for targeting lymphatic system–associated diseases.
Authors
Daisuke Yasuda, Nana Sato, Keisuke Yanagida, Tomomi Hashidate-Yoshida, Tomohiro Shiiya, Hideo Shindou, Atsuki Taira, Takashi Ebihara, Takao Shimizu, Masanori Hirashima, Seiya Mizuno, Satoru Takahashi, Satoshi Ishii
×Abstract
Liver invasion is one of the most frequent events in the progression of gallbladder cancer (GBC). However, the cellular and pathological role of the tumor-liver–interface microenvironment in liver invasion is still enigmatic. Here, we applied single-cell and spatial transcriptomics to systematically investigate the cellular component and gene expression regulation of the microenvironment from the tumor to the liver, specifically the invasive boundary. Our analyses revealed that CXCL9+ macrophage–rich immune cell niches were accumulated in the tumor-liver invasive margin, where 2 subclasses of the CXCL9+ immune cell niches, CXCL9+TRAC+ (CT) and CXCL9+C1QB+ (CC) niches, were identified. CD8+ T cells were recruited by CXCL9+ macrophages through CXCL9-CXCR3 interaction in the CT niche, which was located adjacent to the liver. Moreover, the CC niche was proximal to the tumor core, where tumor cells induced CD8+ T cell exhaustion via LGALS4 expression. In addition, our cohort study showed that high CXCL9 and low LGALS4 in the liver invasion margin demonstrated a favorable prognosis and better responses to anti–PD-1 immunotherapy for patients with gallbladder cancer. Altogether, these findings demonstrate novel cellular and molecular mechanisms underlying liver invasion and offer clinical value for immunotherapies.
Authors
Maolan Li, Zhaonan Liu, Shenbing Shan, Ziyao Jia, Yongsheng Li, Fatao Liu, Lina Lu, Shimei Qiu, Chen Li, Ziyi Wang, Siyuan Yan, Yuhao Zhao, Lili Gao, Zhiqing Yuan, Yuanding Liu, Jiyao Ma, Jiayi Feng, Pengxiao Geng, Yiming Li, Xiaojing Xu, Xinhua Lin, Changjun Liu, Zebing Liu, Wenguang Wu, Xiangsong Wu, Wei Gong, Yanjing Li, Dongxi Xiang, Yongning He, Yun Liu, Rong Shao, Kwan Man, Wu Wei, Yingbin Liu
×Abstract
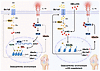
The immune system is not only essential for host defense, but it is also involved in tissue maintenance and disease pathogenesis. Macrophages play a key role in tissue repair, fibrosis, and tumorigenesis, but the mechanisms underlying their multifunctionality have not been fully explored. Here, we identified Mrep (Ly6ChiCX3CR1loPDPN+CD9+) as a crucial subset of macrophages for muscle regeneration after muscle injury. Muscle regeneration required Mrep-derived activin A, which was produced via the TLR4/TIR domain–containing adapter-inducing interferon-β/TANK-binding kinase 1/interferon regulatory factor 3/7 signaling pathway in response to muscle injury. Mrep exerted pathological effects by secreting activin A in a model of genetically induced heterotopic ossification (HO), which was suppressed by TLR4 inhibition. Thus, this study elucidates the context-dependent functions of macrophages and the link between injury and HO, suggesting that Mrep is a potential therapeutic target for regenerating muscles and suppressing HO.
Authors
Wenqiang Yin, Kazuo Okamoto, Asuka Terashima, Warunee Pluemsakunthai, Takehito Ono, Taku Ito-Kureha, Shizuo Akira, Yoshinobu Hashizume, Roland Baron, Satoshi Ueha, Kouji Matsushima, Martin M. Matzuk, Yuji Mishina, Hiroshi Takayanagi
×Abstract
Epidermal growth factor receptor–activating (EGFR-activating) mutations are established biomarkers of resistance to immune checkpoint blockade (ICB) in lung cancer, yet the precise molecular mechanism and effective therapeutic strategies remain elusive. In this study, we show that EGFR overexpression and amplification recapitulated the negative effect of EGFR driver mutations on the ICB response, indicating a proactive involvement of EGFR signaling in antagonizing the antitumor immune response. Functional studies unveiled that EGFR activation suppressed the cellular response to IFN-γ following ICB treatment across multiple cancer models. This impairment in IFN-γ responsiveness further limited the upregulation of T cell–recruiting chemokines and antigen presentation, resulting in reduced T cell infiltration and activation, ultimately undermining antitumor immunity. Mechanistically, EGFR promotes Src homology 2-containing protein tyrosine phosphatase 2 (SHP2) activation to accelerate STAT1 dephosphorylation, leading to premature termination of the IFN-γ response. SHP2 inhibition restored ICB sensitivity in EGFR-activated tumors, significantly reducing tumor burden while maintaining a favorable safety profile. Our findings suggest that the EGFR/SHP2 axis functions as a molecular brake to disrupt the initiation and amplification of the IFN-γ–mediated antitumor response during immunotherapy. This discovery unveils a potential avenue to overcome immunotherapy resistance in EGFR-driven tumors, particularly lung cancer, through SHP2-targeted combination strategies.
Authors
Wei-Tao Zhuang, Lan-Lan Pang, Li-Yang Hu, Jun Liao, Jian-Hua Zhan, Ting Li, Ri-Xin Chen, Jia-Ni Zheng, An-Lin Li, Wen-Yan Yu, Tian-Qin Mao, Liang Chen, Yu-Jian Huang, Shao-Dong Hong, Jing Li, Jun-Han Wu, Yi-Ming Zeng, Meng-Juan Yang, Hai-Qing Zeng, Ya-Xiong Zhang, Li Zhang, Wen-Feng Fang
×Abstract
Surgical stress, such as hepatic ischemia-reperfusion (I/R) injury, induces excessive inflammation and adversely affects liver surgery outcomes. Regulatory T cells (Tregs) are crucial for immune homeostasis, yet their protective mechanisms against liver I/R injury remain unclear. In this study, we demonstrated that decreased hepatic Treg abundance correlates with increased liver injury in patients undergoing hepatic hemangioma resections. In murine models, Treg depletion worsened liver I/R injury. Bulk RNA-seq of hepatic Tregs showed enrichment of Toll-like receptor (TLR) signaling pathways, with flow cytometry identifying TLR4 as the most increased TLR after I/R. Treg-specific Tlr4 knockout mice (Treg-Tlr4–/– mice) exhibited exacerbated liver injury following I/R. Adoptive transfer of WT Tregs, but not Tlr4-deficient Tregs, alleviated liver injury in both Treg-depleted and Treg-Tlr4–/– mice. Transcriptomic analysis revealed that IL-10 production was impaired in Tlr4-deficient Tregs. Mechanistically, Tlr4-deficient Tregs showed reduced activation of the MyD88/ERK/CREB pathway, resulting in diminished IL-10 production. Myd88–/– and IL-10–/– Tregs failed to confer protection against liver I/R injury, whereas exogenous IL-10 administration rescued the hepatic dysfunction in Treg-Tlr4–/– mice. Our findings implicate the vital role of TLR4 in Tregs to mitigate liver I/R injury and offer a potential therapeutic option to reduce postoperative complications following liver surgery.
Authors
Hongji Zhang, Yunwei Zhang, Tianxing Ren, Carolyn Tsung, Peng Song, Peng Xu, Guoliang Wang, Chunyan Cao, Changyan Wang, Ping Sun, Qi Zhang, Yanhong Zhu, Xin Zhong, Yong Guan, Xiaofei Zhang, Han Wang, Jinxiang Zhang, Hui Wang
×Abstract
Dysregulation of cell cycle checkpoints is a cancer hallmark, with ubiquitination-controlled protein stability playing a pivotal role. Although p21, a key cyclin-dependent kinase inhibitor, is tightly regulated by ubiquitin-mediated degradation, the key upstream modulators of its ubiquitination remain incompletely defined. Here, we identify poly(ADP-ribose) glycohydrolase (PARG) as a regulator of p21 stability in gastric cancer (GC) cells. We show that PARG expression is markedly upregulated in GC tissues and correlates with poor patient prognosis. Functional assays revealed that genetic depletion of PARG triggers G2/M phase arrest and impairs GC cell proliferation. Mechanistically, we demonstrate that PARG loss enhances p21 PARylation, which disrupts its association with E3 ubiquitin ligase, thereby reducing K48-linked ubiquitination and leading to p21 protein stabilization. Moreover, we identify lysine residues K161 and K163 as critical sites for PARG-mediated regulation of p21 ubiquitination. Our findings reveal a posttranslational regulatory axis in which PARG governs cell cycle progression by modulating the PARylation-dependent ubiquitination of p21. These results broaden the understanding of p21 regulation in cancer and highlight PARG as a potential therapeutic target for GC treatment.
Authors
Yangchan Hu, Qimei Bao, Yixing Huang, Yan Wang, Xin Zhao, Junjun Nan, Yuxin Meng, Mingcong Deng, Yuancong Li, Zirui Zhuang, Hanyi He, Dan Zu, Yuke Zhong, Chunkai Zhang, Bing Wang, Ran Li, Yanhua He, Qihan Wang, Min Liu, John A. Tainer, Yin Shi, Xiangdong Cheng, Ji Jing, Zu Ye
×Abstract
Glioblastomas (GBMs) are highly lethal brain tumors with limited treatment options and resistance to immune checkpoint inhibitors due to their immunosuppressive tumor microenvironment. Here, we identify OLIG2 as a key regulator of immune evasion in GBM stem-like cells, which inhibits CD8+ T cell–dependent antitumor immunity while promoting protumor macrophage polarization. Mechanistically, OLIG2 recruited HDAC7 to repress CXCL10 transcription, inducing STAT3 activation in tumor-associated macrophages (TAMs) and decreasing CD8+ T cell infiltration and activation. Genetic deletion of OLIG2 significantly increased CXCL10 secretion, shifting TAMs toward an antitumor phenotype and enhancing CD8+ T cell activities. Furthermore, upregulated OLIG2 expression was correlated with resistance to immune checkpoint inhibitors in patients with GBMs. OLIG2 inhibition by either genetic deficiency or pharmacological targeting with CT-179 sensitized GBM tumors to anti–PD-L1 therapy, enhancing antitumor immune responses and prolonging survival. Our findings reveal OLIG2+ glioma stem-like cells as critical mediators of immune evasion and identify the OLIG2/HDAC7/CXCL10 axis as a potential therapeutic target to enhance immune checkpoint inhibitor efficacy and improve immunotherapy outcomes in aggressive GBMs.
Authors
Xinchun Zhang, Jinjiang Xue, Cunyan Zhao, Chenqiuyue Zeng, Jiacheng Zhong, Gangfeng Yu, Xi Yang, Yao Ling, Dazhen Li, Jiaxiao Yang, Yun Xiu, Hongda Li, Shiyuan Hong, Liangjun Qiao, Song Chen, Q. Richard Lu, Yaqi Deng, Zhaohua Tang, Fanghui Lu
×Abstract
Patients with malignant peripheral nerve sheath tumors (MPNSTs) have poor outcomes despite multimodal treatment with surgery, radiation, and systemic therapy. The responses to radiotherapy (RT) are mixed, and the biologic mechanisms underlying this heterogeneity in the radiation response of MPNSTs are not understood. Here, we combined bulk and single-cell transcriptomics, genome-wide CRISPR interference screens, and multiplatform molecular analysis across MPNST cells, mouse allograft models, and patients’ samples to understand the mediators of the radiation response. Our data revealed that MPNSTs, but not benign plexiform neurofibromas, induced a type I IFN signature that functionally mediated the radiation response. Moreover, irradiation of immunocompetent mouse MPNST allografts led to IFN-mediated T cell recruitment and activation. Both host mouse T cells and intact tumor IFN receptor signaling were required for RT’s efficacy in mouse MPNST allografts. Analysis of human MPNST resection specimens demonstrated that increased microenvironmental and CD8+ T cell infiltration were associated with improved local control following RT. These results provide a preclinical rationale for combining immunomodulatory agents targeting IFN signaling to improve radiation responses in MPNSTs and potentially other soft tissue sarcomas.
Authors
Iowis Zhu, Julian Chien, Gabriel E. Rech, Kanish Mirchia, Sixuan Pan, Kaeli Miller, Joanna Pak, Rosanna Wustrack, Varun Monga, Steve E. Braunstein, Mark D. Adams, Line Jacques, Melike Pekmezci, S. John Liu, Harish N. Vasudevan
×Abstract
Atopic dermatitis (AD) is a chronic inflammatory skin condition characterized by a type 2 immune response that is not fully understood. Single-cell RNA-seq of human AD skin and murine models of type 2 inflammation identified transcriptionally distinct fibroblast clusters, revealing IL-4Rα–dependent populations of immune-acting fibroblasts (IAFs). These unbiased findings prompted further investigation into the role of dermal fibroblasts during allergic inflammation. These studies demonstrated that, in an inflammatory environment including TNF-α, IL-1β, and IL-17A, the cytokines IL-4 and IL-13 stimulated both mouse and human fibroblasts to produce multiple chemokines, including CCL8, which activated CCR3 to attract T cells. In the skin, fibroblasts were the primary source of many of these chemokines, and targeted deletion of IL-4Rα in mouse fibroblasts reduced T cell infiltration in a mouse model of AD. Additionally, pharmacologic inhibition of CCR3, the receptor shared by many chemokines produced by fibroblasts, decreased T cell infiltration and skin inflammation in mouse models of AD. These findings demonstrate that dermal fibroblasts are more than passive structural cells; they actively participate in the type 2 immune response and contribute to AD by producing chemokines that increase inflammation. Targeting the functions of IAFs could offer an alternative therapeutic approach for AD.
Authors
Tomofumi Numata, Michael Shia, Yoshiyuki Nakamura, Fengwu Li, Hung Chan, Teruaki Nakatsuji, Kellen J. Cavagnero, Jared Simmons, Henry Li, Aaroh Anand Joshi, Marta Palomo-Irigoyen, Richard L. Gallo
×Abstract
Lymphatics maintain fluid homeostasis, immune surveillance, and tissue integrity. Here, we identified the E26 transformation-specific transcription factors Erg and Fli1 as essential cooperative regulators of lymphatic integrity and function. Using inducible lymphatic endothelial cell–specific deletion in mice, we demonstrated that combined loss of Erg and Fli1 in adults results in fatal lymphatic failure, including chylothorax, chylous ascites, and impaired lymphatic drainage. Single-cell transcriptomic analysis revealed that loss of Erg and Fli1 causes disrupted lymphatic heterogeneity and dysregulation of key lymphatic genes, including valve-specific gene profiles. Erg and Fli1 coordinated lymphatic-immune crosstalk by transcriptionally regulating C-C motif chemokine ligand 21, which mediates DC trafficking. Erg or Fli1 loss also induced proinflammatory and prothrombotic gene expression, further contributing to lymphatic dysfunction. During embryonic development, the codeletion led to lymphatic mispatterning and loss of valve-initiating lymphatic endothelial cell clusters. The impact of loss of Erg and Fli1 function on lymphatic development in mice is consistent with FOXC2 mutations in lymphedema-distichiasis syndrome or ERG gene variants underlying primary lymphedema in humans. Moreover, Erg and Fli1 were required for regenerative lymphangiogenesis and lymphatic repair following injury in adults. Our findings establish Erg and Fli1 as core transcriptional regulators of lymphatic identity, integrity, and function.
Authors
Myung Jin Yang, Seok Kang, Seon Pyo Hong, Hokyung Jin, Jin-Hui Yoon, Cheolhwa Jin, Chae Min Yuk, Lydia Getachew Gebeyehu, Junho Jung, Sung-Hwan Yoon, Hyuek Jong Lee, Gou Young Koh
×Abstract
N-acetyl-l-leucine (NALL), a derivative of the branched-chain amino acid leucine, has shown therapeutic potential for neurodegenerative diseases, including in prodromal stages of Parkinson’s disease (PD). However, the mechanism of its protective effects has been largely unknown. Using human induced pluripotent stem cell–derived dopaminergic neurons from patients carrying GBA1, LRRK2, or VPS35 mutations, as well as from sporadic PD cases, we found that NALL treatment markedly reduced Ser129 phosphorylated α-synuclein (pS129-syn). Discovery-based proteomic analysis revealed that NALL treatment upregulated lysosomal, mitochondrial, and synaptic proteins without inducing cytotoxicity. The reduction of pS129-syn was dependent on serine protease HTRA1, which was robustly induced by NALL. Moreover, NALL increased the expression of wild-type parkin in mutant dopaminergic neurons, leading to increased glycosylated dopamine transporter, elevated synaptic membrane-associated synaptojanin-1, and accelerated synaptic vesicle endocytosis, suggesting improved synaptic function. Furthermore, in LRRK2R1441C knockin mice, NALL administration decreased pS129-syn, elevated parkin levels, and ameliorated dopamine-dependent motor learning deficits. These findings highlight the therapeutic potential of NALL for PD by its protective effects on α-synuclein pathology and synaptic function in vulnerable dopaminergic neurons.
Authors
Pingping Song, Chuyu Chen, Rossella Franchini, Bryan Duong, Yi-Zhi Wang, Robert Coukos, Zhong Xie, Jeffrey N. Savas, Yueqin Zhou, Mariarita Bertoldi, D. James Surmeier, Loukia Parisiadou, Dimitri Krainc
×Abstract
BACKGROUND Kidney stone disease (KSD) affects approximately 10% of the population. While genetic factors are known to play a role in KSD, determining the clinical relevance of rare variants in KSD genes identified in adults remains challenging.METHODS The Swiss Kidney Stone Cohort is a multicenter longitudinal, observational study consisting of kidney stone formers (KSFs) (n = 701) and non-kidney stone formers (NKSFs) (n = 200). Blood and urine samples were collected at enrollment and over 3 years for deep biochemical phenotyping. Results were correlated with rare genetic variants in established KSD genes identified through whole-exome sequencing and classified according to American College of Medical Genetics and Genomics and the Association of Molecular Pathology (ACMG/AMP) criteria.RESULTS Collectively, we found rare (likely) pathogenic (LP/P) variants representing strong KSD risk factors in 6.8% of KSFs, predominantly in genes involved in renal phosphate handling and cystinuria. Detailed biochemical analyses confirmed that KSFs carrying heterozygous LP/P SLC34A3 variants exhibited significant hyperphosphaturia. In contrast, monoallelic LP/P variants in SLC34A1, SLC9A3R1, or CYP24A1, which were also frequent in NKSFs, did not result in the expected biochemical alterations, calling into question their causative role as strong KSD risk factors. In cystinuria, monoallelic SLC7A9 variants represented intermediate risk factors, since they caused biochemical alterations but required additional factors for KSD occurrence, based on frequent LP/P variants in NKSFs. The presence of strong risk factors was associated with higher kidney stone (KS) recurrence over the 3-year observation period, supporting a predictive value for genetic testing.CONCLUSIONS Correlation of genetic findings with thorough biochemical phenotyping and comparison with NKSFs redefines the clinical relevance of variants in KSD genes and has prognostic value.
Authors
Johannes Münch, Jana Petrovska, Joana Figueiro-Silva, Isabel Rubio-Aliaga, Elena M. Cabello, Ivan Ivanovski, Michael Papik, Beatrice Oneda, Daniel G. Fuster, Harald Seeger, Thomas Ernandez, Florian Buchkremer, Gregoire Wuerzner, Nasser A. Dhayat, Alexander Ritter, Stephan Segerer, Beat Roth, Anita Rauch, Pietro Manuel Ferraro, Olivier Bonny, Carsten A. Wagner, Ruxandra Bachmann-Gagescu
×Abstract
Malignant tumors with TP53 mutations exhibit poor therapeutic outcomes and high recurrence rates. T cell receptor–based (TCR-based) T cell therapy shows great promise for targeting intracellular cancer neoantigens. However, the immunogenic potential of TP53 hotspot mutations remains poorly characterized. Here, we identified an immunogenic neoantigen derived from the recurrent TP53R248Q mutation, presented by the prevalent HLA-A*11:01 allele. Additionally, we isolated a TP53R248Q-reactive TCR that specifically recognized the TP53R248Q mutation without any discernible cross-activity with cognate WT TP53 or other TP53 mutants at the same codon position. Functional characterization revealed that TP53R248Q TCR-T cells exhibited selective cytotoxicity against tumor cells expressing both the TP53R248Q mutation and HLA-A*11:01 in vitro. Importantly, the adoptive transfer of TP53R248Q TCR-T cells exhibited significant antitumor activity in a clinically relevant patient-derived xenograft model engrafted with TP53R248Q/HLA-A*11:01–positive human tumor tissues. Collectively, our study validates the immunogenicity of the TP53R248Q hotspot mutation and provides a TCR with high therapeutic potential for the development of T cell therapies targeting TP53R248Q/HLA-A*11:01–positive cancers.
Authors
Lianghua Shen, Ziyu Chen, Jian Xu, Qiaomei He, Changmeng Zhang, Xiao Zhou, Xiaodan Ding, Jinan Fang, Fanlin Li, Ming Jiao, Yuqin Yang, Baoxia Dong, Liping Wan, Xueying Ding, Yan Zheng, Jingyi Zhou, Chijian Zuo, Tian Min, Ming Zhu, Bin Ma, Yuhua Wan, Qiufang Guo, Hua Zhang, Jian Hua, Pengran Wang, Qi Li, Jiang Long, Xianmin Song, Yan Zhang
×Abstract
Neuroinflammation, encompassing both innate and adaptive immune responses, plays a crucial role in ischemic stroke. Although B lymphocytes are central to adaptive immunity, their contributions to ischemic stroke remain poorly understood. Here, we demonstrated that B lymphocytes accumulate in ischemic lesions, forming germinal center–like structures at the later stage after stroke, which mainly depended on in situ proliferation. This accumulation correlated with worsened neuroinflammation and ischemic injury, whereas B cell depletion reduced chronic brain damage during stroke. Mechanistically, microglia recruited B cells into ischemic lesions through MIF-CD74/CXCR4 signaling during the early phase of stroke, while IFN-related pathways in B cells further drove neuroinflammation and brain injury. Targeting these pathways markedly alleviated cerebral ischemia and inflammation. Our findings shed light on the role of B lymphocytes in stroke pathology and suggest promising new avenues for therapeutic intervention.
Authors
Sheng Yang, Hang Zhang, Lu-Lu Xu, Luo-Qi Zhou, Yun-Hui Chu, Lian Chen, Xiao-Wei Pang, Lu-Yang Zhang, Li-Fang Zhu, Ming-Hao Dong, Ke Shang, Jun Xiao, Long-Jun Wu, Wei Wang, Dai-Shi Tian, Chuan Qin
×Abstract
Drug-associated environmental cues can trigger drug-seeking behavior and precipitate relapse. In this study, we determined that the claustrum (CL) connects the ventral tegmental area (VTA) with the medial prefrontal cortex (mPFC), forming the VTA–CL–mPFC circuit. Using a methamphetamine (METH) conditioned place preference (CPP) model in male mice, we found that manipulating the VTA–CL–mPFC circuit or CL neuronal ensemble receiving projections from VTA and projecting to mPFC (VTA–CL–mPFC) could disrupt the retrieval of METH-paired context memory, resulting in the blockage of the acquisition of METH CPP in male mice. During the process, dopamine release and dopamine 1-like receptor–mediated activation of CL neurons were required for the retrieval of METH-induced reward memory in male mice. These findings reveal a midbrain–cortical circuit orchestrated by CL neurons that plays an essential role in the retrieval of drug-paired environmental cue memory.
Authors
Ziheng Zhao, Yuhong He, Yang Liu, Quying Feng, Hee Young Kim, Yu Fan, Xiaowei Guan
×Abstract
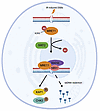
While current antivirals primarily target viral proteins, host-directed strategies remain underexplored. Here, we performed a genome-wide CRISPR inhibition (CRISPRi) screening to identify the host protein, hepatocyte growth factor-regulated tyrosine kinase substrate (HGS), facilitating the pan-coronavirus infection both in vitro and in vivo. Mechanistically, HGS interacts with the viral membrane (M) protein, facilitating its trafficking to the ER-Golgi intermediate compartment for virion assembly. Conversely, HGS deficiency caused M retention in the ER, blocking assembly. Leveraging this interaction, we designed M-derived peptides and screened over 5,000 FDA-approved or commonly used drugs, identifying riboflavin tetrabutyrate (RTB). Both the peptides and RTB bind HGS and disrupt its interaction with the M protein, leading to M retention in the ER and subsequent blockade of virion assembly. These agents demonstrated broad anti-pan-coronavirus activity in vitro and in vivo. Collectively, our findings establish HGS as a druggable host target and identify RTB as a promising broad-spectrum antiviral candidate.
Authors
Xubing Long, Rongrong Chen, Rong Bai, Buyun Tian, Yu Cao, Kangying Chen, Fuyu Li, Yiliang Wang, Yongjie Tang, Qi Yang, Liping Ma, Fan Wang, Maoge Zhou, Xianjie Qiu, Yongzhi Lu, Jie Zheng, Peng Zhou, Xinwen Chen, Qian Liu, Xuepeng Wei, Yongxia Shi, Yanhong Xue, Jincun Zhao, Wei Ji, Liqiao Hu, Jinsai Shang, Tao Xu, Zonghong Li
×Abstract
BACKGROUND Amino acid (AA) concentrations are increased in prediabetes and diabetes. Since AAs stimulate glucagon secretion, which should then increase hepatic AA catabolism, it has been hypothesized that hepatic resistance (associated with hepatic fat content) to glucagon’s actions on AA metabolism leads to hyperglucagonemia and hyperglycemia.METHODS To test this hypothesis, we therefore studied lean and obese individuals, the latter group with and without hepatic steatosis as defined by proton density fat fraction (PDFF) > 5%. After an overnight fast, femoral vein, femoral artery, and hepatic vein catheters were placed. [3-3H] glucose and l-[1-13C,15N]-leucine were used to measure glucose turnover and leucine oxidation, respectively. During a hyperglycemic clamp, an AA mixture was infused together with insulin and glucagon (1.5 ng/kg/min 0–120 minutes; 3.0 ng/kg/min 120–240 minutes). Tracer-based measurement of hepatic leucine oxidation in response to rising glucagon concentrations and splanchnic balance (measured using arteriovenous differences across the liver) of the other AAs were the main outcomes measured.RESULTS The presence of hepatic steatosis did not alter hepatic glucose metabolism and leucine oxidation in response to insulin and rising concentrations of glucagon. Splanchnic balance of a few AAs and related metabolites differed among the groups. However, across-group differences of AA splanchnic balance in response to glucagon were unaffected by the presence of hepatic steatosis.CONCLUSION The action of glucagon on hepatic AA metabolism is unaffected by hepatic steatosis in humans.TRIAL REGISTRATION Clinical Trials.gov: NCT05500586.FUNDING NIH National Institute of Diabetes and Digestive and Kidney Diseases DK116231, DK78646, DK116231, DK126206, and DK116231.
Authors
Hannah E. Christie, Sneha Mohan, Aoife M. Egan, Federica Boscolo, Chiara Dalla Man, Scott M. Thompson, Michael Jundt, Chad J. Fleming, James C. Andrews, Kent R. Bailey, Michael D. Jensen, K. Sree Nair, Adrian Vella
×Abstract
Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome is an immunodeficiency caused by autosomal dominant hyperfunctional mutations in chemokine receptor CXCR4 that promote panleukopenia due to BM retention. We previously reported a preclinical gene therapy protocol involving allele-nonspecific Cxcr4 CRISPR/Cas9 inactivation, leveraging the known in vivo dominance of Cxcr4+/o (+, WT; o, inactivated) hematopoietic stem cells (HSCs) for autologous BM engraftment and leukocyte reconstitution over HSCs with other Cxcr4 genotypes. Here, we show that without BM conditioning, this approach is not able to correct leukopenia in WHIM mice. We therefore modified the protocol by adding conditioning with a nongenotoxic CD117-targeted immunotoxin, CD117-antibody-saporin-conjugate. With this change, donor-derived blood cells rapidly reached ~95% chimerism after transplantation, which was stable without adverse events. Mice receiving edited HSCs showed rapid normalization of absolute myeloid cell counts, the key blood subset responsible for WHIM syndrome. In competitive transplants using equal numbers of edited and unedited donor HSCs, over 80% of blood cells originated from the edited population, predominantly with the Cxcr4+/o genotype. These results provide proof of principle that CRISPR/Cas9-mediated inactivation of the Cxcr4 disease allele, combined with nongenotoxic HSC-targeted conditioning, may offer a safe and effective gene therapy strategy generalizable to all WHIM-causing mutations.
Authors
Ji-Liang Gao, Zhanzhuo Li, Rafael Calderon-Perez, Antonia Pavek, Lina Kim, David H. McDermott, Philip M. Murphy









In-Press Preview - More
Abstract
BACKGROUND. Estrogen deficiency and progressive hearing loss (HL) are significant concerns in individuals with Turner syndrome (TS). However, whether childhood estrogen deficiency increases HL risk and whether estrogen replacement therapy (ERT) prevents hearing deterioration are still unclear. METHODS. This prospective cohort study recruited children with TS from a tertiary referral center between 2016 and 2024. All participants received standardized recombinant human growth hormone therapy. Longitudinal monitoring data of hormone levels, metabolic parameters, and annual audiological examinations were recorded. The primary analysis used a multivariate Cox model to estimate the adjusted hazard ratio of hearing loss between estrogen-deficient and estrogen-normal TS patients without prior exogenous estrogen exposure. The secondary analysis compared annual pure tone average (PTA) and its changes between the ERT and non-ERT groups in a substudy. RESULTS. Among 87 prepubertal pediatric TS patients, 48 (55.2%) were estrogen-deficient, 38 HL events occurred over 35-month median follow-up. The estrogen-deficient group had higher HL incidence (27 cases, 56.3%; 20.6/100 person-years [PY]) versus estrogen-normal (11 cases, 28.2%; 8.6/100 PY), with estrogen deficiency independently increasing HL risk (HR = 2.93; 95%CI:1.21–7.12). Notably, estrogen deficiency also independently predicted abnormal DPOAE with an even higher effect size (HR = 3.98, 95% CI: 1.35–11.76). The substudy found that initiating ERT at age of 12 significantly preserve auditory function, with the ERT group showing markedly lower PTA and slower hearing deterioration (–1.24 dB/y vs. 1.13 dB/y right ear; –1.85 dB/y vs. 1.04 dB/y left ear, P = 0.001). CONCLUSION. Childhood estrogen deficiency is a modifiable risk factor. Initiating ERT around early adolescence may help hearing preservation. TRIAL REGISTRATION. ChiCTR2300068063. FUNDING. National Natural Science Foundation of China (82173154 and 82471155), Fundamental Research Funds for the Central Universities, Clinical Research 5010 Program, Sun Yat-sen University: 2024004.
Authors
Yan Huang, Liyang Liang, Yanfang Ye, Lina Zhang, Li Ling, Zhe Meng, Wei Liu, Jia Guo, Zulin Liu, Zhen Zhao, Zhigang Zhang, Yu Si
Abstract
Isolating commensal fungi from mouse intestines has been challenging, limiting our understanding of their role in intestinal immune homeostasis and diseases. Using an Fc fusion protein of the C-type lectin Dectin-2, we successfully purified the commensal Ascomycota fungus Engyodontium sp. from mouse feces. Engyodontium enhances the antimicrobial activity of colonic neutrophils via CARD9 pathway, and exacerbates colitis by impairing the colonization of intestinal Lactobacillus johnsonii (L. johnsonii) WXY strain. L. johnsonii produces high levels of L-glutamic acid by expressing the glutaminase-encoding gene glsA to facilitate Treg expansion via enhancing IL-2 receptor signaling. Patients with Crohn’s disease (CD) and ulcerative colitis harbored increased Engyodontium and decreased L. johnsonii abundance. Engyodontium directly induced calprotectin in human colonic neutrophils, and CD patients exhibited lower levels of L-glutamic acid which also promoted human Treg expansion. These findings highlight the Engyodontium-calprotectin axis against the Lactobacillus-glutamate-Treg cascade to aggravate colitis, suggesting commensal Engyodontium-triggered signaling as a therapeutic target for mucosal inflammatory diseases.
Authors
Xinying Wang, Haiyang Sun, Ying Tan, Shaoting Xu, Zishan Liu, Kaile Ji, Ding Qiu, Jianping Deng, Bingbing Feng, Xueting Wu, Yoichiro Iwakura, Minhu Chen, Rui Feng, Chanyan Huang, Ce Tang
Abstract
Osteofibrous dysplasia (OFD) is a skeletal RASopathy presenting with periosteal bone lesions that may progress to fracture and delayed healing (pseudarthrosis). MET gene mutations reducing ubiquitin-mediated protein degradation via loss of the juxtamembrane domain (METΔJMD) were previously identified in OFD patients, resulting in ligand-dependent gain-of-function. The impact of METΔJMD expression on skeletal progenitor cell differentiation and the potential efficacy of targeted therapies remain unclear. We engineered MetΔJMD mice and showed that MetΔJMD expression inhibited osteogenic differentiation of skeletal progenitor cells in vitro and impaired cortical bone development and reduced bone stiffness in vivo. In contrast, conditional deletion of Met enhanced osteogenic differentiation of periosteal progenitor cells. Inhibition of MAPK signaling with MEK inhibitors restored osteogenic differentiation of mouse MetΔJMD skeletal progenitor cells and promoted activation of transcriptional signatures associated with skeletal development and osteoblast differentiation in OFD patient pseudarthrosis-derived primary cells. With this preclinical support, we treated with the MEK inhibitor mirdametinib a pediatric OFD patient suffering from a 3-year history of persistent pseudarthrosis, resulting in fracture union. Our findings demonstrate a bi-directional role for MET in regulating osteogenic differentiation of skeletal progenitor cells and a therapeutic avenue to improve clinical outcomes for this, and potential other, skeletal RASopathies.
Authors
Aysha Khalid, Kristin Denton, Nandina Paria, Ila Oxendine, Meghan Wassell, Reuel Cornelia, Sasidhar Uppuganti, Jeffry S. Nyman, G. Jayashree Jagadeesh, Carlos R. Ferreira, Simon J. Conway, Robert E. Hammer, John O. Ritter, Mylinh Nguyen, David A. Podeszwa, Laura J. Klesse, Carol A. Wise, Jonathan J. Rios
Abstract
Metabolic syndrome and excessive alcohol consumption (MetALD) result in liver injury and fibrosis, which is driven by increased collagen production by activated hepatic stellate cells (HSCs). Our previous studies demonstrated that LARP6, an RNA-binding protein, may facilitate collagen production. However, the expression and function of LARP6 as a regulator of fibrosis development in a disease-relevant model remain poorly understood. We demonstrated that LARP6 was upregulated in human activated HSCs in metabolic dysfunction-associated steatohepatitis (MASH) and MetALD. By using snRNA/ATAC-sequencing, we showed that JUNB upregulated LARP6 expression in activated HSCs. Moreover, LARP6 knockdown in human HSCs suppressed fibrogenic gene expression. By integrating eCLIP analysis and ribosome profiling in HSCs, we showed that LARP6 interacted with mature mRNAs comprising over 300 genes, including RNA structural elements within COL1A1, COL1A2, and COL3A1 to regulate mRNA expression and translation. IP-MS analysis demonstrated LARP6 protein–protein interactions with mRNA translation components and the actin cytoskeleton. Furthermore, dsiRNA-based HSC-specific gene knockdown or pharmacological inhibition of LARP6 attenuated fibrosis development in human MASH and MetALD liver spheroids. Our results suggest LARP6 plays a key role in fibrogenic gene regulation and that targeting LARP6 in human HSCs may represent a therapeutic approach for liver fibrosis.
Authors
Hyun Young Kim, Orel Mizrahi, Wonseok Lee, Sara B. Rosenthal, Cuijuan Han, Brian A. Yee, Steven M. Blue, Jesiel Diaz, Jyotiprakash P. Jonnalagadda, Lena A. Street, Kanani Hokutan, Haeum Jang, Charlene Miciano, Chen-Ting Ma, Andrey A. Bobkov, Eduard Sergienko, Michael R. Jackson, Marko Jovanovic, Branko Stefanovic, Tatiana Kisseleva, Gene W. Yeo, David A. Brenner
Abstract
Neonatal life is marked by rapid antigen exposure, necessitating establishment of peripheral immune tolerance via conversion of naïve CD4+ T cells into regulatory T cells (Tregs). Here, we demonstrated heightened capacity for FOXP3 expression and tolerogenic function among cord blood versus adult blood naive CD4+ T cells and showed that this is linked to their unique metabolic profile and elevated expression of the NADase, CD38. Early-life naïve CD4+ T cells demonstrated a metabolic preference for glycolysis, which directly facilitated their differentiation trajectory. We revealed an age-dependent gradient in CD38 levels on naïve CD4+ T cells and showed that high CD38 expression contributes to both the glycolytic state and tolerogenic potential of neonatal CD4+ T cells, effects that were mediated at least in part via the NAD-dependent deacetylase SIRT1. Thus, the early-life window for peripheral tolerance in humans is critically enabled by the immunometabolic state of the naïve CD4+ compartment.
Authors
Laura R. Dwyer, Andrea M. DeRogatis, Sean Clancy, Victoire Gouirand, Charles Chien, Elizabeth E. Rogers, Scott P. Oltman, Laura L. Jelliffe-Pawlowski, Theo van den Broek, Femke van Wijk, Susan V. Lynch, Rachel L. Rutishauser, Allon Wagner, Alexis J. Combes, Tiffany C. Scharschmidt
View more articles by topic:
Sign up for email alerts
Review Series - More
Series edited by Daniel J. Drucker
Clinical innovation and scientific progress in GLP-1 medicine
Series edited by Daniel J. Drucker
Therapies targeting the glucagon-like peptide 1 (GLP-1) receptor have revolutionized the treatment of obesity and diabetes. This series of reviews, curated by Dr. Dan Drucker, describes the latest research in this fast-moving in field, from our evolving understanding of the mechanism of GLP-1 receptor signaling to the medicines’ impact on inflammation and the consequences for heart, kidney, and brain health. The reviews also explore the impact of these medicines on conditions beyond their initial indications, including cancer and neurodegenerative disease risk.
×



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)